2021.06.03.18
Files > Volume 6 > Vol 6 No 3 2021
Código de barras de ADN de tres especies de árboles frutales con potencial económico del valle de Huaura, Lima, Perú
DNA barcoding of three species of fruit trees with economic potential from the Huaura Valley, Lima, Peru

Hermila Belba Díaz-Pillasca , Angel David Hernández-Amasifuen *, Miguel Machahua , Alexandra Jherina Pineda-Lázaro , Alexis Argüelles-Curaca, Brayan Lugo
Available from: http://dx.doi.org/10.21931/RB/2021.06.03.18
RESUMEN
El Perú presenta una gran diversidad de recursos genéticos, pero a la vez se desaprovechan especies por desconocimiento o bajo rendimiento económico. Situación que se refleja en el valle de Huaura con los árboles frutales de cansaboca (Bunchosia armeniaca), palillo (Campomanesia lineatifolia) y naranja agria (Citrus aurantium), especies con gran importancia en la gastronomía tradicional local, pero en la actualidad catalogadas en peligro crítico. Con el fin de conservar estas especies se planteó como objetivo establecer código de barras de ADN de tres especies amenazadas con potencial económico del valle de Huaura. Se extrajo ADN de las tres especies con el método CTAB y para las amplificaciones en PCR se emplearon los cebadores de código de barras de ADN universales pertenecientes a cloroplastos: matK, rbcL y trnH-psbA. A partir de los productos purificados y cuantificados se realizó el secuenciamiento de las muestras. Las secuencias fueron analizadas, alineadas y agrupadas con los programas Bioedit, Codon Code Aligner y MEGA respectivamente. Las concentraciones de ADN fueron: palillo (457 ng/μl), cansaboca (433 ng/μl) y naranja agria (442 ng/μl). La amplificación de los cebadores produjo productos de PCR entre 357 y 810 pb. Las secuencias de NCBI que presentaron mayor porcentaje de identidad con cada especie en estudio fueron sometidas a análisis filogenético, los cuales colocaron a las especies en grupos distintos y revelando diferencia genética con las muestras estudiadas. Se proporcionaron las herramientas básicas para implementar códigos de barras de ADN en tres especies de árboles frutales en el valle de Huaura.
Palabras clave: Bunchosia armeniaca, Campomanesia lineatifolia, Citrus aurantium, palillo, naranja agria.
SUMMARY
Peru has a great diversity of genetic resources, but at the same time, species are wasted due to ignorance or low economic performance. The situation reflected in the Huaura valley with the fruit trees of cansaboca (Bunchosia armeniaca), palillo (Campomanesia lineatifolia) and sour orange (Citrus aurantium), species with great importance in the traditional local gastronomy, but currently classified as endangered critically. To conserve these species, the objective was to establish DNA barcoding of three threatened species with economic potential in the Huaura Valley. DNA from the three species was extracted with the CTAB method, and universal DNA barcode primers belonging to chloroplasts were used for the PCR amplifications: matK, rbcL, and trnH-psbA. Samples were sequenced from the purified and quantified products. The sequences were analyzed, aligned, and grouped with the Bioedit, Codon Code Aligner, and MEGA programs. The DNA concentrations were: cansaboca (457 ng / μl), palillo (433 ng / μl) and sour orange (442 ng / μl). Amplification of the primers produced PCR products between 357 and 810 bp. The NCBI sequences that presented the highest percentage of identity with each species under study were subjected to phylogenetic analysis, which placed the species in different groups and revealed genetic differences with the studied samples. The essential tools were provided to implement DNA barcoding in three species of fruit trees in the Huaura valley.
Keywords: Bunchosia armeniaca, Campomanesia lineatifolia, Citrus aurantium, palillo, sour orange.
INTRODUCCIÓN
El Perú presenta gran riqueza biológica por la alta diversidad genética, debido a esto es considerado como uno de los países más importantes en recursos genéticos1. Estos recursos pueden permitir un mayor desarrollo económico y social, siempre que se incluya una alta responsabilidad con respecto a especies endémicas y rescate de especies domesticadas2. El desaprovechamiento de especies por desconocimiento o bajo rendimiento económico conlleva a la perdida de material genético valioso, situación que en la actualidad se refleja en el valle de Huara, provincia de Huaura, departamento de Lima; con árboles frutales como cansaboca o ciruela del fraile (Bunchosia armeniaca Cav. DC), palillo (Campomanesia lineatifolia Ruiz & Pav.) y naranja agria (Citrus aurantium L.), especies con gran demanda en décadas pasadas e importancia en la gastronomía tradicional local3. Pero en la actualidad existe una baja o nula presencia en mercados locales, mientras que en campos de cultivo es difícil encontrar estas especies, presentándose muy pocos individuos en huertas por herencia familiar, debido a esto son catalogadas en peligro crítico4,5.
Estas especies vegetales, así como muchas especies de otras regiones del país presentan gran potencial socio-económico. Por lo que el Perú, viene desarrollando objetivos estratégicos como parte del Convenio sobre la Diversidad Biológica, de las cuales se ha trazado el país la meta de evitar la extinción de especies mediante el avance de conocimientos, con una gran base científica y tecnológica para aumentar la valoración y funcionalidad de estas especies6. Como parte del avance científico se están desarrollando técnicas avanzadas relacionadas a biología molecular, tales como estudios de código de barras, genómica, proteómica, metabolómica, transcriptómica2.
El código de barras de ADN es un método para identificar las especies de todo tipo de organismo vivo empleando una secuencia corta de ADN (<1000 pb) que evoluciona lo suficientemente rápido como para diferir entre especies estrechamente relacionadas7. Cuando se recupera una secuencia de código de barras de una muestra desconocida, se utiliza un algoritmo para compararla con una base de datos de referencia que contiene códigos de barras de muestras de especies identificadas, lo que permite su comparación y posible identificación de manera rápida y clara6,8,9. En otras palabras, los códigos de barras de ADN funcionan como identificadores moleculares para cada especie, de la misma manera que los códigos de barras en blanco y negro legibles por máquina se utilizan en la industria minorista para identificar productos comerciales10,11,12.
Las secuencias para código de barras que se emplean en las investigaciones están relacionadas a sus alineamientos múltiples y número de variaciones, las cuales permiten distinguir las especies emparentadas sin influir en su asignación correcta a través de la variación intraespecífica13. Estas secuencias de ADN son: ribulosa-bisfosfato carboxilasa (rbcL), maturasa K (matK) y el espaciador intergénico trnH-psbA (trnH-psbA). Cada uno de las secuencias presenta diferencias en sus regiones codificantes, es así como rbcL presenta una región de codificación universal que no evoluciona rápidamente, matK presenta una región de codificación que evoluciona bastante rápido y trnH-psbA es un espaciador intergénico que evoluciona rápidamente14,15.
La identificación molecular de especies mediante código de barras de ADN facilita la selección de material vegetal desde plántulas, permitiendo la conservación de especies amenazadas. Por lo tanto, en la investigación se planteó el objetivo de establecer el código de barras de ADN de tres especies con potencial económico del valle de Huaura, para facilitar su identificación a nivel molecular.
MÉTODOS
Material vegetal y extracción de ADN.
Se emplearon muestras de hojas de las especies Bunchosia armeniaca, Campomanesia lineatifolia y Citrus aurantium, las cuales fueron colectadas de la campiña de Huacho y transportadas al Laboratorio de Biotecnología Vegetal de la Universidad Nacional José Faustino Sánchez Carrión – Huacho. La extracción de ADN se realizó según el protocolo de bromuro de cetiltrimetilamonio (CTAB)16. Se tomaron alrededor de 100 mg de tejido de hojas frescas, se añadieron 2,85 μl de β-mercaptoetanol y 1.000 μl de tampón de extracción CTAB 2% (100 mM Tris, 28 mM EDTA, 2% CTAB, 1,4 M NaCl y 2,5% PVP), las muestras se trituraron de forma mecánica y se incubó a 65º C durante 45 min y. Posteriormente, se añadió 900 μl de cloroformo-alcohol isoamílico (24:1) y se mezcló suavemente durante 5 min. Las muestras se centrifugaron a 14.000 rpm durante 5 min. Después de la centrifugación, se recogió la fase acuosa y se adicionaron 60 μl de CTAB al 10% y se incubó a 65° C por 5 min. Se añadió 900 μl de cloroformo-alcohol isoamílico (24:1) y se mezcló suavemente durante 5 min y centrifugo a 14.000 rpm durante 5 min, se retiró el sobrenadante y se adiciono isopropanol frio en el mismo volumen que el sobrenadante obtenido. Las muestras fueron colocadas a -20° C durante 30 min. Posteriormente, se centrifugó a 14.000 rpm durante 20 min, se decantó el sobrenadante y se invirtieron los tubos sobre papel toalla durante 1 min. Seguidamente, se lavó con etanol al 70% y se centrifugo a 14.000 rpm por 5 min. Se añadió 300 μl de CH3CO2K 5 M frio y se incubó a -20° C durante 10 min. Luego se centrifugó a 14.000 rpm por 5 min, se eliminó el sobrenadante y se realizó un lavado con etanol al 90%. Seguidamente se eliminó el etanol y se invirtieron los tubos sobre papel toalla estéril. Una vez seco el pellet se añadió 100 μl de agua libre de nucleasas, posterior se adicionó 3 μl de RNAsa al 2% y se incubó a 37° C durante 60 min. A continuación, las muestras se centrifugaron a 14.000 rpm durante 5 min y los sedimentos de ADN se lavaron dos veces con etanol al 70% y se invirtieron los tubos sobre papel toalla estéril durante 30 min. Los sedimentos de ADN se diluyeron con 50 µl de agua libre de DNAsa y posteriormente se cuantifico en Nanodrop ND-1000 (NanoDrop Technologies, USA) y estimo la calidad mediante electroforesis en gel de agarosa al 1%. El ADN extraído se almacenó a -20 ° C hasta su posterior uso.
Amplificación por PCR y secuenciamiento.
Se emplearon los siguientes cebadores de código de barras de ADN universales pertenecientes a cloroplastos: matK, rbcL y trnH-psbA (Tabla 1). Las reacciones de PCR se realizaron en un volumen de 25 μl que contenía 12.5 μl master mix (10 mM Tris HCl, 50 mM KCl, 2 mM de MgCl2, 0,2 mM de dNTP, 0,02 U/μl de Taq Polimerasa), 0.5 μl de cada cebador (forward y reverse), 1 μl de ADN genómico y 10,5 μl de agua pura estéril.
Se utilizaron los siguientes parámetros del termociclador: desnaturalización inicial de 94° C durante 5 min, seguido de 40 ciclos a 94° C durante 30 s, hibridación a 52° C para los cebadores rbcL y matK durante 30 s, 55° C para cebadores de trnH-psbA durante 30 s y extensión a 72° C durante 1 min, seguido de una extensión final a 72° C por 10 min. Las amplificaciones se realizaron en el termociclador PCRMax (BioSystems, UK).
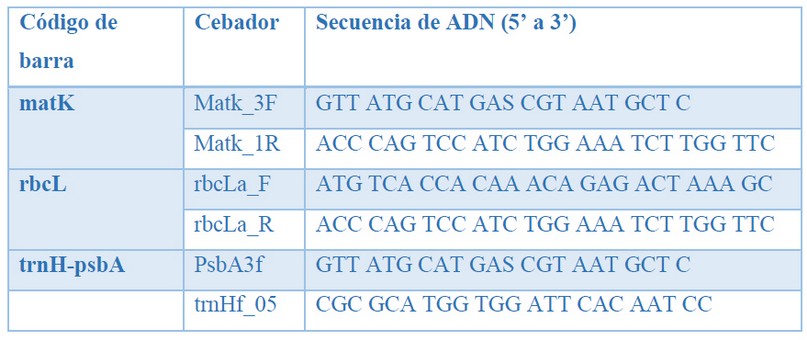
Tabla 1. Secuencia de cebadores para código de barras de ADN.
Los productos de amplificación por PCR se sometieron a electroforesis en geles de agarosa al 1% durante 30 minutos a 40 V, detectados por tinción con azul de bromofenol, los resultados se observaron bajo luz ultravioleta (UV) mediante un sistema de documentación de geles con transiluminador UV (Illuminyx, USA). El tamaño de los productos de PCR se determinó utilizando DNA ladder de 100 pb (Promega, Madison). Los productos de PCR purificados se cuantificaron usando un espectrofotómetro Nanodrop ND-1000 (NanoDrop Technologies, USA). El secuenciamiento de los productos de PCR se realizó en la empresa MACROGEN (Corea del Sur).
Análisis de datos.
Las secuencias obtenidas fueron analizadas con el programa Bioedit y alineadas con el programa Codon Code Aligner. La identificación de códigos de barras desconocidos a partir de hojas se realizó básicamente mediante datos de la Herramienta de búsqueda de alineación local básica (BLAST) con un corte mínimo de 98% de identidad para una coincidencia superior7. Estos resultados se verificaron mediante agrupación y análisis filogenéticos en los que comparamos las ramas de especímenes desconocidos con secuencias de especies de referencia, para ello las secuencias de ADN se alinearon y curaron empleando el algoritmo MUSCLE del programa MEGA 7.0. Los árboles filogenéticos se construyeron en MEGA 7.0 utilizando el método de Neighbor-Joining (NJ) con 1.000 bootstraps basado en el modelo de parámetros de Kimura 217. Se construyeron árboles filogenéticos con los fragmentos combinados.
RESULTADOS
Los resultados obtenidos permitieron establecer que, la técnica de CTAB fue eficiente para la extracción de ADN en las tres especies; lo cual se comprobó con la verificación de la integridad del ADN extraído. La relación de absorbancia 260/280 fue cercana a 1,80, lo que significa ADN altamente puro para el análisis de códigos de barras. Las concentraciones de ADN en diferentes accesiones fueron: Bunchosia armeniaca (457 ng/μl), Campomanesia lineatifolia (433 ng/μl) y Citrus aurantium (442 ng/μl). La amplificación exitosa de los tres cebadores de códigos de barras produjo productos de PCR entre 300 y 800 pb (Figura 1).

Figura 1. Productos de PCR amplificados con los cebadores trnH-psbA en las tres especies en peligro de extinción del valle de Huaura. La posición de las muestras en el gel de agarosa (1-2) Campomanesia lineatifolia, (3-4) Bunchosia armeniaca, (5-6) Citrus aurantium y (-) control negativo; M: DNA ladder de 100 pb para la determinación del tamaño de las muestras.
Las secuencias de nucleótidos obtenidas mostraron diferencias en longitud y contenido de guanina-citocina (GC) en las muestras analizadas con cada cebador de código de barras (Tabla 2). En el caso del cebador matK, la longitud de secuencia más alta de Bunchosia armeniaca (810 pb). Se observó una variación de longitud de secuencia en Citrus aurantium (357 pb) con el cebador matK. La longitud media de la secuencia de rbcL fue casi uniforme (573 pb). Citrus aurantium presento la menor longitud (373 pb) y contenido de GC (26,27 %) con el cebador trnH-psbA.
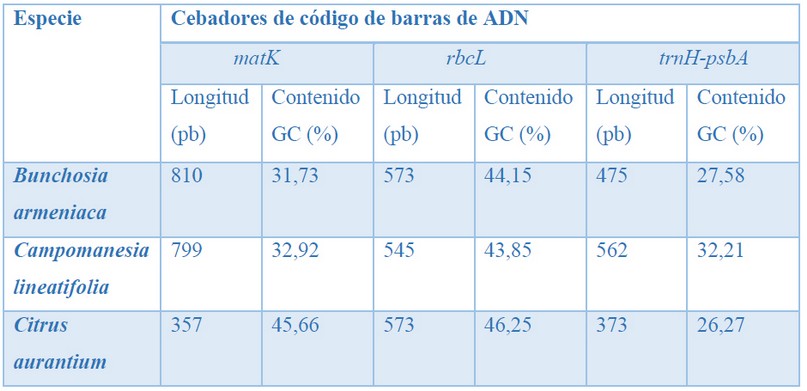
Tabla 2. Características de secuencia de nucleótidos de marcadores de código de barras de ADN.
Las secuencias obtenidas de este estudio se compararon con el banco de datos de ADN publicados en BLAST de NCBI (Tabla 3, 4 y 5), presentando en algunos casos una identidad superior al 98% con las secuencias NCBI de especies de plantas conocidas y podrían estar emparentadas a ellas.
Las especies que han logrado presentar mayor porcentaje de identidad con Bunchosia armeniaca del valle de Huaura, se encontraron en el rango de 99,75% hasta 98,28% de identidad (Tabla 3), manteniendo una baja variabilidad entres especies del mismo género. Las comparaciones de Campomanesia lineatifolia muestran un descenso en el porcentaje de identidad con las demás especies, mientras se van alejando del género, pero manteniendo en común la familia (Myrtaceae) (Tabla 4). En el caso de Citrus aurantium se logró encontrar porcentajes de identidad de 99,83% hasta 99,47%, dentro del mismo género con 99% de cubierta de consulta (Tabla 5), así mismo dentro de la lista se presentan tres accesiones de Citrus aurantium de otras partes del mundo, mostrándose que no comparten tantos porcentajes de identidad debido posiblemente a una evolución necesaria al habitad de cada región.
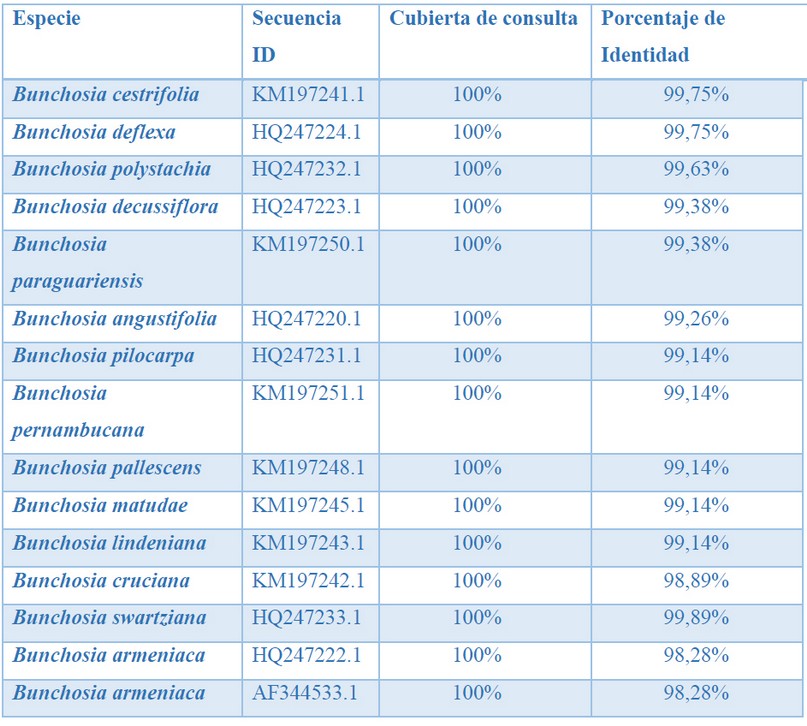
Tabla 3. Especies de mayor homología en BLAST con muestras de Bunchosia armeniaca del valle de Huaura.
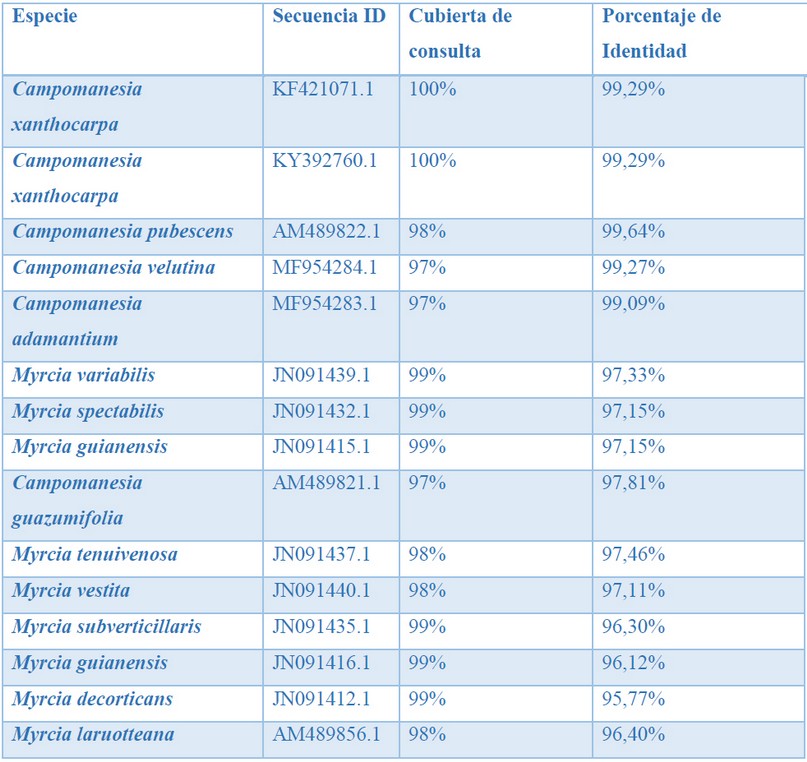
Tabla 4. Especies de mayor homología en BLAST con muestras de Campomanesia lineatifolia del valle de Huaura.

Tabla 5. Especies de mayor homología en BLAST con muestras de Citrus aurantium del valle de Huaura.
Las secuencias de NCBI que presentaron mayor porcentaje de identidad con cada especie en estudio (Campomanesia lineatifolia, Bunchosia armeniaca y Citrus aurantium) fueron sometidas a análisis filogenético.
La alineación combinada de las secuencias de las especies en estudio (Figura 2) con secuencias de 13 (Citrus aurantium) y 15 (Bunchosia armeniaca y Campomanesia lineatifolia) especies de mayor porcentaje de similitud y cubierta de consulta, descargadas de NCBI mostró una variabilidad las posiciones de nucleótidos. De manera similar, se construyó un árbol filogenético combinado de máxima verosimilitud mediante la concatenación de datos de secuencia de tres cebadores de códigos de barras (matK, rbcL y trnH-psbA) para estimar las divergencias evolutivas entre cada especie en estudio con las especies de mayor porcentaje de identidad descargadas de NCBI.
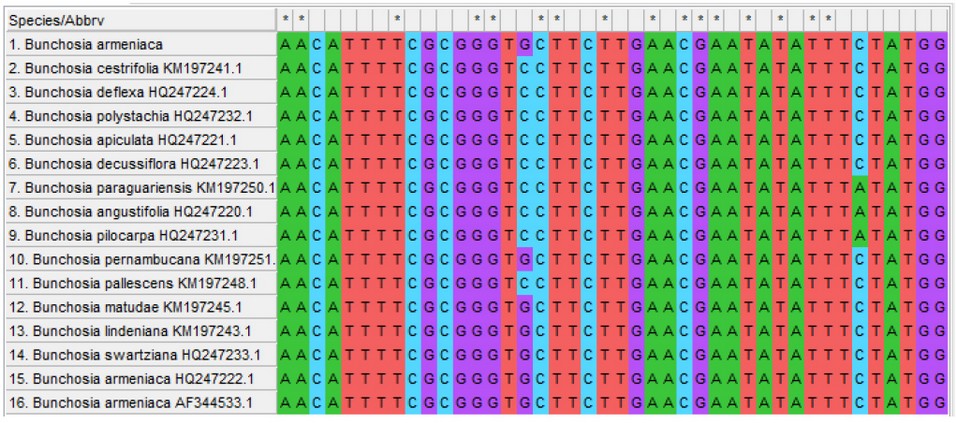
Figura 2. Alineamiento combinado de las secuencias con el programa MEGA.
En el árbol filogenético de Bunchosia armeniaca (Figura 3) se reveló que la muestra en estudio es genéticamente diferente a las especies de referencia determinadas por BLAST y se colocan en grupos distintos. Se consideró secuencias de la misma especie (Bunchosia armeniaca) localizadas en México y se mostró una diferencia genética con la muestra estudiada en Huaura, Perú.
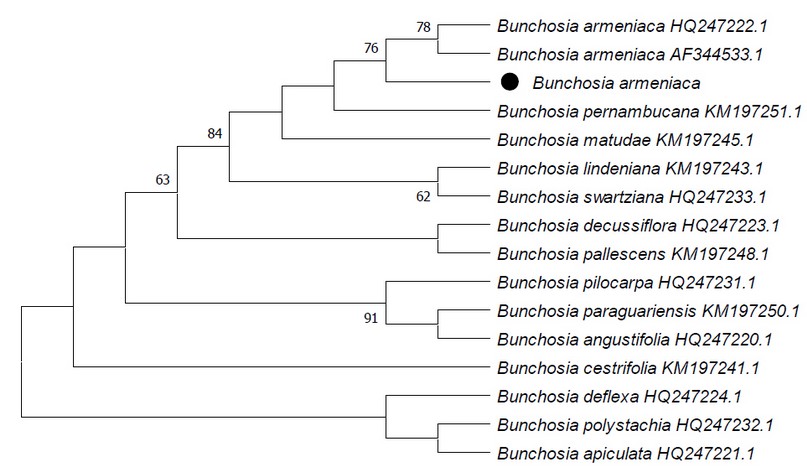
Figura 3. Árbol filogenético de máxima probabilidad de evolución divergente en Bunchosia armeniaca del valle de Huaura.
En el caso de Campomanesia lineatifolia no se encontró alguna secuencia de ADN de la misma especie en BLAST, por lo que se presenta los primeros resultados de secuenciamiento relacionada a esta especie. El árbol filogenético revelo que se encuentra muy emparentado con Campomanesia xanthocarpa, agrupados en un solo clado con 100% de soporte de arranque (Figura 4).
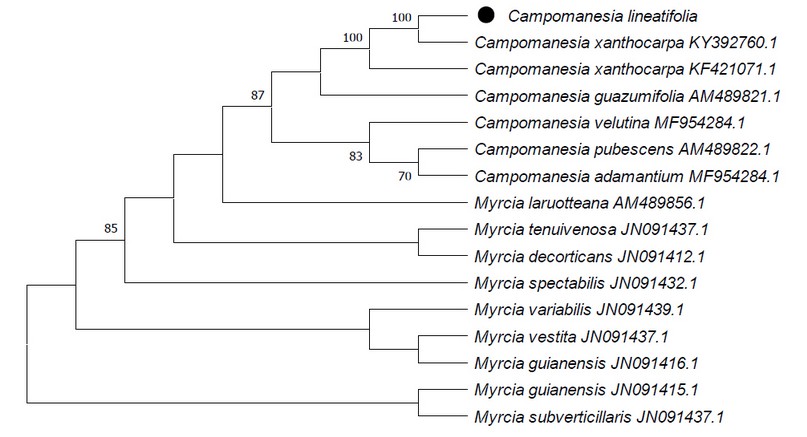
Figura 4. Árbol filogenético de máxima probabilidad de evolución divergente en Campomanesia lineatifolia del valle de Huaura.
Mientras que el árbol filogenético de la naranja agria reveló una diferencia genética con las muestras de Citrus aurantium de referencia determinadas por BLAST situadas en Canadá, Estados Unidos e Italia, colocándose en grupos separados y distintos. Con la muestra de naranja agria de referencia de Grecia se revelo una menor diferencia al estar en un grupo cercano con 87% de soporte de arranque (Figura 5).
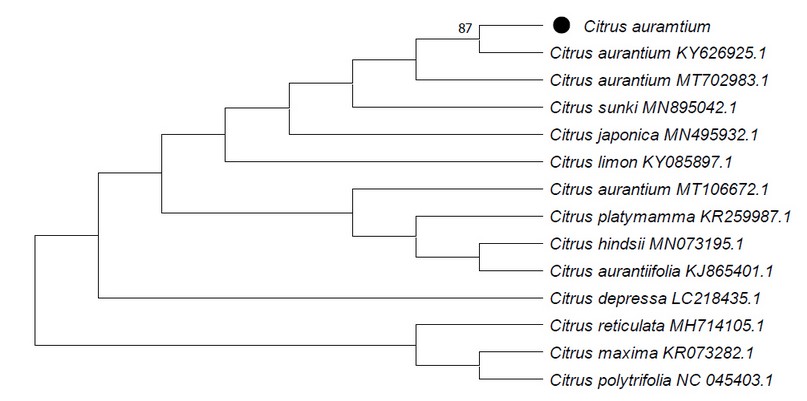
Figura 5. Árbol filogenético de máxima probabilidad de evolución divergente en Citrus aurantium del valle de Huaura.
DISCUSIÓN
Se ha aplicado con éxito un método que utiliza secuencias cortas de ADN, conocidas como "códigos de barras de ADN", para identificar especies de plantas, especialmente loci en el genoma de los plástidos como matK, rbcL, y trnH-psbA8,13.
La evaluación de la aplicabilidad universal mediante la cuantificación por PCR y el éxito de la secuencia es el primer paso para determinar la idoneidad de un fragmento de ADN dado como código de barras. A este respecto, todas las regiones analizadas (matK, rbcL y trnH-psbA) se amplificaron de manera efectiva, lo que permitió una secuenciación simple y de alta calidad. Por lo que, los cebadores empleados en las tres especies en estudio presentaron características ideales para código de barras de ADN tales como capacidad de amplificación, alta eficiencia de amplificación y secuenciación, también variación genética suficientemente alta para distinguir secuencias a nivel de especie, pero también suficientemente conservadora entre individuos de la misma especie6,18.
La amplificación de la región trnH-psbA fue exitosa, con amplicones más cortos (aproximadamente 570 pb) que permitió una secuenciación efectiva, al ser fácilmente amplificable usando cebadores universales y sus niveles de discriminación de especies son generalmente más altos que para otras regiones, considerándose un marcador valioso8,19,20,21. Los resultados obtenidos presentan similitud con otros grupos de plantas, donde la amplificación de la región trnH-psbA y la calidad de secuenciación fue lo suficientemente alta como para considerarlo un código de barras7,21,22. Pero algunos grupos taxonómicos presentan limitaciones que han reducido severamente la utilidad de trnH-psbA como código de barras. Su alta frecuencia de variación de longitud resultante de sus numerosas inserciones y eliminaciones dificulta la construcción de una alineación precisa de estas secuencias8.
En el presente estudio Bunchosia armeniaca del valle de Huara se separó en otro grupo con Bunchosia armeniaca de referencia de México, esto podría demostrar una combinación de dispersión temprana de larga distancia entre América del Sur y México, aunque no podemos determinar la ubicación geográfica precisa donde estos clados comenzaron a diversificarse. Pero esto podría relacionarse al predominio de la dispersión en la formación de un grupo de especies regionales23. El marcador matK presento 810 pb de longitud y 31,73% de contenido GC, mostrando el marcador con mayor longitud en comparación con rbcL y trnH-psbA¸ pero presenta variaciones entre especies respeto a su amplificación y longitud; es por ello que el marcador matK presenta división entre investigadores respecto a su universalidad como cebador. Presentándose estudios donde se indican que este marcador es clave en el código de barras de ADN, al discriminar grupos específicos11,24. Pero también están otros autores que cuestionan la utilidad de este gen como código de barras debido a la pobre amplificación y secuenciación. eficiencia y problemas relacionados con la universalidad de los cebadores25,26.
Campomanesia lineatifolia no ha presentado antecedentes en relación al secuenciamiento de su cloroplasto, como si encontraron de Campomanesia xanthocarpa secuencias parciales con el espaciador intergénico trnH-psbA, con una longitud de 569 pb y con menos del 1% de gaps27. Con la secuencia de matK se obtuvo una longitud de 799 pb, la misma longitud obtenida por (28) empleando la misma secuencia en Campomanesia xanthocarpa; mientras que en el mismo estudio empleando rbcL obtuvieron una longitud mayor (645 pb) que la del presente estudio (546 pb), así mismo se presenta mucha similitud con las tres secuencias con 99% de identidad, a partir de las investigaciones realizadas en Campomanesia xanthocarpa de Brasil. Demostrando que la diversificación es claramente un contribuyente importante a la riqueza de especies existentes en esta región. La secuencia que fue más conservadora (entre las tres regiones analizadas) en términos de variabilidad molecular fue rbcL, proporcionando una amplia información para la selección de cebadores específicos de orden apropiado. Esto indica que rbcL tiene el menor número de sitios variables y que la secuencia de cebador seleccionada tiene una fuerte universalidad. Por lo tanto, es recomendable rbcL como un fragmento efectivo para el código de barras de ADN en especies arbóreas. Esto también fue confirmado por (6) y otros autores11,19, quienes recomiendan rbcL y matK para el código de barras de ADN central para plantas.
El espaciador intergénico trnH-psbA en Citrus aurantium de la presente investigación presento una menor longitud en comparación a Citrus aurantium de referencias de otras partes del mundo, pero con la secuencia rbcL se obtuvo la misma longitud, llegando a presentar una identidad de casi 100%29,30. Finalmente, con la secuencia de matK no se presentaron identidades dentro del género Citrus. La separación de grupos de esta misma especie podría estar relacionada a la diversificación general de especies introducidas de manera individual permitiendo explicar el patrón de dispersión frecuente y continua de nuevos linajes, esto plantea la posibilidad de que la caracterización de grupos de especies esté dominada por linajes únicos que se trasladaron a una región, lo que parece haber brindado a individuos el tiempo necesario para adaptarse a nuevas condiciones abióticas23. Así mismo la alta frecuencia de las variantes de longitud de trnH-psbA en algunas especies impactó la identificación de especies utilizando métodos basados en la alineación, así como la comparación con las especies del presente estudio mediante el análisis de distancia, que tuvo un efecto mínimo en las búsquedas de BLAST8,20.
El análisis filogenético a partir del código de barras de ADN permite mejorar la identificación de especies, al proporcionar caracteres adicionales que pueden estar conducidas por variaciones variación intraespecífica e inversiones interespecíficas que pueden confundir las relaciones entre especies estrechamente relacionadas31,32. Permitiendo identificar a las especies exactas que se encuentra en peligro de extinción en el valle de Huaura, y, además, mostrando un distanciamiento entre la misma especie ubicada en otras partes del mundo.
De esta manera se puede emplear estos métodos como base de cualquier estudio científico de identificación completamente perfecta de material vegetal, conjuntamente con la enorme biblioteca digital en línea de códigos de barras de ADN, que sirve como información para buscar muestras (no identificadas o identificadas) utilizando uno o unos pocos loci de códigos de barras de ADN20.
CONCLUSIONES
El presente estudio es el primero en analizar especies frutales con potencial económico en peligro de extinción del valle de Huaura para facilitar la identificación a nivel molecular. Nuestros resultados proporcionan las herramientas básicas para implementar códigos de barras de ADN en especies nativas en el valle de Huaura, y pueden ayudar a elegir un código de barras de ADN apropiado para aplicaciones de alta productividad. Dichas aplicaciones de alto rendimiento podrían mejorar en gran medida los protocolos de monitoreo de la biodiversidad que se utilizan para estudiar la ecología y conservación de especies en peligro de extinción.
AGRADECIMIENTOS
A la Universidad Nacional José Faustino Sánchez Carrión, por el financiamiento del estudio mediante fondos del área prioritaria FOCAM: Preservación de la biodiversidad, identificación y desarrollo de las potencialidades económicas de la zona de impacto del Proyecto de Camisea; con Resolución Vicerrectoral N° 0044-2013-VTR-UNJFSC. Así como al Blgo. Fernando Serna y Blgo. Alfredo Martin por sus sugerencias en la culminación de la investigación.
REFERENCIAS
- León B, Pitman N, Roque J. Introducción a las plantas endémicas del Perú. Revista Peruana de Biología 2006, 13(2): 9-22. DOI: https://doi.org/10.15381/rpb.v13i2.1782
- von May R, Catenazzi A, Angulo A, Venegas P, Aguilar C. Investigación y conservación de la biodiversidad en Perú: importancia del uso de técnicas modernas y procedimientos administrativos eficientes. Revista Peruana de Biología 2012, 19(3): 351-358. DOI: https://doi.org/10.15381/rpb.v19i3.1055
- Ministerio del Ambiente (MINAM). Estrategia Nacional de Diversidad Biológica al 2021 y su Plan de acción 2014-5018. Ministerio del Ambiente, Dirección General de Diversidad Biológica. Lima, Perú. 2014. Consultado 19 jul. 2020. Disponible en https://sinia.minam.gob.pe/documentos/estrategia-nacional-diversidad-biologica-2021-plan-accion-2014-2018
- Mostacero J, Mejía F, Gastañadui D, De La Cruz J. Inventario taxonómico, fitogreográfico y etnobotánico de frutales nativos del norte del Perú. Scientia Agropecuaria 2017, 8(3): 215-224. DOI: http://dx.doi.org/10.17268/sci.agropecu.2017.03.04
- Díaz H, Honorio Z, Hernández A, Durand M, Gózalo A, Domínguez G. Identificación de especies frutícolas nativas con potencialidad productiva en peligro de extinción en la provincia de Huaura. Aporte Santiaguino 2020, 13(2): 78-91. DOI: https://doi.org/10.32911/as.2020.v13.n2.700
- CBOL Plant Working Group. A DNA barcode for land plants. Proceedings of the National Academy of Sciences of the United States of America 2009, 106: 12794-12797. DOI: https://doi.org/10.1073/pnas.0905845106
- Kress WJ, García-Robledo C, Uriarte M, Erickson DL. DNA barcodes for ecology, evolution, and conservation. Trends in Ecology & Evolution 2015, 30(1): 25-35. DOI: https://doi.org/10.1016/j.tree.2014.10.008
- Kress JW, Wurdack KJ, Zimmer EA, Weigt LA, Janzen DH. Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences 2005, 102(23): 8369-8374. DOI: https://doi.org/10.1073/pnas.0503123102
- Little DP, Stevenson DW. A comparison of algorithms for the identification of specimens using DNA barcodes: examples from gymnosperms. Cladistics 2007, 23(1): 1–21. DOI: https://doi.org/10.1111/j.1096-0031.2006.00126.x
- Lahaye R, van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G, Maurin O, Duthoit S, Barraclough TG, Savolainen V. DNA barcoding the floras of biodiversity hotspots. Proceedings of the National Academy of Sciences 2008, 105(8): 2923-2928. DOI: https://doi.org/10.1073/pnas.0709936105
- Newmaster SG, Grguric M, Shanmughanandhan D, Ramalingam S, Ragupathy S. DNA barcoding detects contamination and substitution in North American herbal products. BMC Medicine 2013, 11(1): 222. DOI: https://doi.org/10.1186/1741-7015-11-222
- Michel CI, Meyer RS, Taveras Y, Molina J. The nuclear internal transcribed spacer (ITS2) as a practical plant DNA barcode for herbal medicines. Journal of Applied Research on Medicinal and Aromatic Plants 2016, 3(3): 94–100. DOI: https://doi.org/10.1016/j.jarmap.2016.02.002
- Hollingsworth PM, Graham SW, Little DP. Choosing and using a plant DNA barcode. PLoS One 2011, 6: e19254. https://doi.org/10.1371/journal.pone.0019254
- Group CP, Hollingsworth PM, Forrest LL, Spouge JL, Hajibabaei M, Ratnasingham S, van der Bank M, Chase MW, Cowan RS, Erickson DL. A DNA barcode for land plants. Proceedings of the National Academy of Sciences 2009, 106(31): 12794-12797. DOI: https://doi.org/10.1073/pnas.0905845106
- de Groot GA, During HJ, Maas JW, Schneider H, Vogel JC, Erkens RH. Use of rbcL and trnL-F as a two-locus DNA barcode for identification of NW-European ferns: an ecological perspective. PLoS One 2011, 6(1): e16371. DOI: https://doi.org/10.1371/journal.pone.0016371
- Doyle JJ, Doyle JL. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochemical Bulletin 1987, 19: 11-15.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Molecular Biology and Evolution 2018, 35:1547-1549. DOI: https://doi.org/10.1093/molbev/msy096
- Cowan RS, Chase MW, Kress WJ, Savolainen V. 300 000 species to identify: problems, progress, and prospects in DNA barcoding of land plants. Taxon 2006, 55(3): 611–616. DOI: https://doi.org/10.2307/25065638
- Bieniek W, Mizianty M, Szklarczyk M. Sequence variation at the three chloroplast loci (matK, rbcL, trnH-psbA) in the Triticeaetribe (Poaceae): comments on the relationships and utility in DNA barcoding of selected species. Plant Systematics and Evolution 2015, 301(4):1275-1286. DOI: https://doi.org/10.1007/s00606-014-1138-1
- Kress WJ, Erickson DL. DNA barcodes: genes, genomics, and bioinformatics. Proceedings of the National Academy of Sciences 2008, 105(8): 2761–2762. DOI: https://doi.org/10.1073/pnas.0800476105
- Pang XH, Song JY, Zhu YJ, Xu HX, Huang LF, Chen SL. Applying plant DNA barcodes for Rosaceae species identification. Cladistics 2010, 27(2): 165–170. DOI: https://doi.org/10.1111/j.1096-0031.2010.00328.x
- Pang XH, Liu C, Shi LC, Liu R, Liang D, Li H, Cherny S, Chen S. Utility of the trnH–psbA intergenic spacer region and its combinations as plant DNA barcodes: a metaanalysis. PLoS ONE 2012, 7(11): e48833. DOI: https://doi.org/10.1371/journal.pone.0048833
- Willis C, Franzone B, Xi Z, Davis C. The establishment of Central American migratory corridors and the biogeographic origins of seasonally dry tropical forests in Mexico. Frontiers in Genetics 2014, 5:433. DOI: http://dx.doi.org/10.3389/fgene.2014.00433
- De Mattia F, Bruni I, Galimberti A, Cattaneo F, Casiraghi M, Labra M. A comparative study of different DNA barcoding markers for the identification of some members of Lamiaceae. Food Research International 2011, 44: 693-702. DOI: https://doi.org/10.1016/j.foodres.2010.12.032
- Du ZY, Qimike A, Yang CF, Chen JM, Wang Q. Testing four barcoding markers for species identification of Potamogetonaceae. Journal of Systematic and Evolution 2011, 49(3):246-251. DOI: https://doi.org/10.1111/j.1759-6831.2011.00131.x
- Yan HF, Hao G, Hu CM, Ge XJ. DNA Barcoding in closely related species: A case study of Primula L. sect. Proliferae Pax (Primulaceae) in China. Journal of Systematics and Evolution 2011, 49: 225-236. DOI: http://dx.doi.org/10.1111/j.1759-6831.2011.00115.x
- Bolson M, Smidt-Ede C, Brotto ML, Silva-Pereira V. ITS and trnH-psbA as Efficient DNA Barcodes to Identify Threatened Commercial Woody Angiosperms from Southern Brazilian Atlantic Rainforests. PLoS One 2015, 10(12): e0143049. DOI: https://doi.org/10.1371/journal.pone.0143049
- Lima RA, Oliveira AA, Colletta GD, Flores TB, Coelho RL, Días P, Frey GP, Iribar A, Rodrigues RR, Souza VC, Chave J. Can plant DNA barcoding be implemented in species-rich tropical regions? A perspective from São Paulo State, Brazil. Genetics and Molecular Biology 2018, 41(3): 661-670. DOI: https://doi.org/10.1590/1678-4685-GMB-2017-0282
- Kress WJ, Erickson DL. A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2007, 2(6): e508. DOI: https://doi.org/10.1371/journal.pone.0000508
- Cornara L, Borghesi B, Canali C, Andrenacci M, Basso M, Federici S, Labra M. Smart drugs: green shuttle or real drug?. Int J Legal Med. 2013, 127(6): 1109-1123. DOI: https://doi.org/10.1007/s00414-013-0893-9.
- Whitlock BA, Hale AM, Groff PA. Intraspecific inversions pose a challenge for the trnH-psbA plant DNA barcode. PLoS ONE 2010, 5(7): e11533. DOI: https://doi.org/10.1371/journal.pone.0011533
- Degtjareva GV, Logacheva MD, Samigullin TH, Terentieva EI, Valiejo-Roman CM. Organization of chloroplast psbA-trnH intergenic spacer in dicotyledonous angiosperms of the family Umbelliferae. Biochemistry (Moscow) 2012, 77(9): 1056-1064. DOI: https://doi.org/10.1134/S0006297912090131
Received: 21 April 2021
Accepted: 20 June 2021
Hermila Belba Díaz-Pillasca , Angel David Hernández-Amasifuen *, Miguel Machahua , Alexandra Jherina Pineda-Lázaro , Alexis Argüelles-Curaca, Brayan Lugo
Universidad Nacional José Faustino Sánchez Carrión, Facultad de Ciencias, Laboratorio de Biotecnología Vegetal, Huacho, Perú,