2023.08.02.62
Files > Volume 8 > Vol 8 No 2 2023
Cytogenetic, genomic and interactome evaluation in an unusual case of a patient with XX/XY chimerism, hermaphroditism and association with situs inversus totalis

César Paz-y-Miño1, Juan Luis García2, Jesús María Hernández-Rivas2, Paola E. Leone3
1 Faculty of Health Sciences Eugenio Espejo, Universidad UTE. Quito, Ecuador.
2 Biomedical Research Institute of Salamanca – Institute of Health Sciences Studies of Castilla y León. Salamanca, Spain.
3 Laboratory of Genetics and Genomics. Sociedad de Lucha contra el Cáncer, SOLCA. Quito, Ecuador.
Correspondence to: [email protected]
Available from: http://dx.doi.org/10.21931/RB/2023.08.02.62
ABSTRACT
We present a scarce case in which three pathologies converge: XX/XY chimerism, hermaphroditism with ovotestis and situs inversus totalis. She is a patient of mixed ethnicity, with predominantly Amerindian markers, who underwent a conventional cytogenetic study, FISH and DNA study of peripheral blood (mesoderm) and buccal mucosa scraping (ectoderm), additionally X-rays, hormonal tests and biopsy of the right gonad. The patient presents hypogonadism, female hypoplastic external sexual organs, and Tanner sexual development 1-2. X-rays show situs inversus totalis. The data for female and male hormones are altered. The karyotype showed the presence of two cell populations, XX and XY, confirmed with FISH and a study of the positive SRY gene. The evaluation of the DNA by numerical Arrays verified the presence of chromosomes X and Y; in the same way, gains and losses of essential genes related to the patient's phenotype were evidenced. We performed an in silico evaluation of the interactome of proteins involved in the clinical manifestations that reflect the alterations of the corresponding genes, which allowed us to make a genotype-phenotype correlation. In conclusion, the studies with a complete clinical, laboratory and genetic panel allowed us to define its alterations.
Keywords: chimerism, 46, XX/46, XY, ovotestis, situs inversus totalis
INTRODUCTION
In 1 in 8,000 to 25,000 individuals1, 2, there may be a change in the position of the organs, placing them on the opposite side, which forms a mirror image called situs inversus. It was first described in 1824, although X-rays confirmed it in 18973. Situs inversus is a rare genetic malformation that can affect one or all human body organs (Situs inversus totalis). It is associated with dextrocardia, including great vessels, and in some cases, complete dextro position of visors and internal organs1. The etiology of this anomaly is complex and is associated, among other reasons, with the mutation of some genes that involve laterality and ciliary movements2, 4-6.
Human chimeras are rare and are usually identified only after birth. Most of the described chimeras have a normal female or male phenotype7, 8, and less than 50% have been reported with an XX/XY9-12 karyotype. As far as we have been able to investigate, there are no reports of patients with situs inversus totalis associated with chimerism, that is, the presence in an individual with more than one genetically differentiated cell population originating from two or more zygotes10, 12. It has been described rarely; true XX/XY hermaphroditism is considered chimeras13-16.
Two X chromosomes determine the formation of ovaries, and the chromosome combination X with Y determines the formation of testicles. Disorders of sexual development (DSD) are defined as congenital conditions in which the development of chromosomal, gonadal or anatomical sex is atypical17, 18. Within the DSD is true hermaphroditism, a rare alteration that presents ovarian and testicular tissue, in the same individual, with variable phenotypes: female, intersex or male; some 200 cases have been described15. Intersex patients may have variable clinical features17. Similarly, chromosome studies show mostly XX karyotypes, XX/XY and fewer XY8,13, 15.
We present a case with cytogenetic and molecular studies by array analysis in two types of tissues, blood and buccal mucosa, with an unusual and rare association between situs inversus totalis and true XX/XY hermaphroditism with clear indicators of chimerism.
CASE PRESENTATION
The present case is an 11-year-old patient, an indigenous ethnic group, product of a third normal pregnancy and cephalic delivery, APGAR score 9-10, low weight (1,200 g). She has no pathological family history of genetic interest or another personal pathological history. Patient evaluations meet all bioethical and informed consent requirements.
She currently presents a particular phenotype: short stature (1.35m <p10), broad shoulders, narrow hips, he impresses as hypogonadal; round face, broad midface, smallmouth, retrognathia, short neck, decreased sexual development (Tanner 2-3) (Figures 1A, 1B and 1C); hands with tiny fingers and mild clinodactyly of the 5th finger (Figures 1D and 1E). On auscultation and palpation, the heart sound was found to be on the right side, as were the liver and spleen. Breast development was found, the appearance of short pubic and axillary hair, and the examination of the external sexual organs showed hypoplastic labia, a small clitoris, and no menstrual cycles had started.
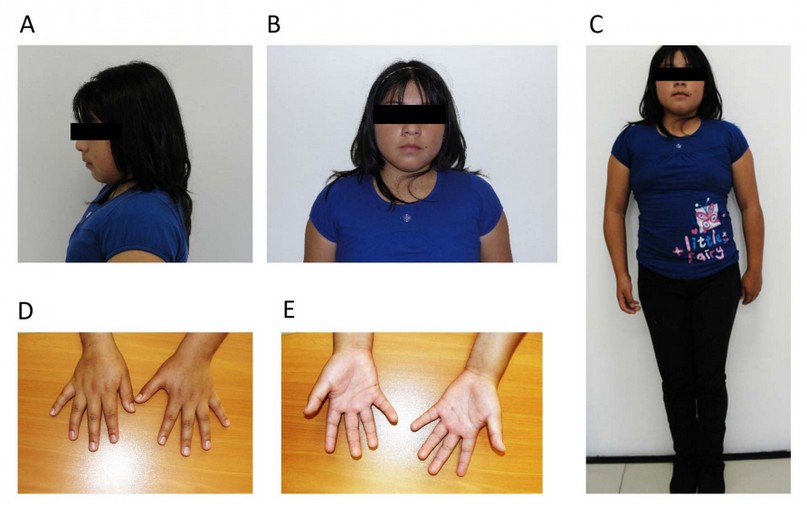
Figure 1. Photographs of the patient. A) Side view. B) Front view. C) Full-length frontal view. D) Palms down. E) Palms up.
A chest X-ray, pelvic abdominal and gonadal ultrasound, and hormonal studies of estradiol, follicle-stimulating, testosterone and corticosteroids were requested. The simple chest X-ray study showed situs inversus totalis (Figures 2A and 2B). The pelvic sonogram showed an incipient uterus in anteverted flexion, hypoplastic tubes, and both gonads in the pelvic cavity. The histopathological study of the biopsy of the right gonad reported the presence of mixed ovarian and testicular tissue compatible with ovotestis. The results for estradiol 32 pg/ml, FSH 2 mUL/ml and testosterone 150 nmol/l show hormonal dysfunction.

Figure 2. X-ray of the patient shows dextrocardia. A) Thorax. B) Abdomen and pelvis.
Genetic study
The proband and her parents underwent a cytogenetic study with the conventional technique. 72-hour culture of whole heparinized blood in RPMI-1640 medium, supplemented with 15% fetal bovine serum, phytohemagglutinin, antibiotics, and antifungals. Harvest with colchicine, hypotonic shock (ClK), and Carnoy's fixative. Chromosome staining with Giemsa and GTG (Trypsin Giemsa) chromosome banding at 450 resolution bands. One hundred metaphases were evaluated.
FISH (fluorescent in situ hybridization) technique was performed, with specific probes for the centromeres of the X chromosome (CEP X) and Y chromosome (CEP Y). Slides were hybridized with 7 ul of hybridization buffer, 1 ul of CEP X (Alpha satellite DNA: Xp11.1-q11.1) Spectrum Green DNA probe, 1 ul of CEP Y (Alpha satellite DNA: Yp11.1-q11. 1) Spectrum Orange DNA probe and 1 ul of water. The Vysis protocol (Abbott, IL, USA) was followed.
DNA was extracted from peripheral blood and buccal mucosa samples for molecular studies. Peripheral blood samples were obtained by venipuncture and treated with ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. 200 µl of the sample were used for DNA extraction using the commercial Purelink Genomic DNA kit (Invitrogen®, Life Technologies, CA, USA). DNA was extracted from the patient's saliva with the Abyntek® Genotex Orange OG-510 DNA Kit. The concentration and purity of the genetic material were assessed using the NanoDrop 2000c Spectrophotometer (Thermo Fisher Scientific Inc., MA, USA).
DNA extracted from peripheral blood was used to study ancestry and analyze the SRY gene. Determination of genetic ancestry was performed with 46 insertion- and deletion-based ancestry informative markers19.
For the amplification of the SRY gene, the previously described standard primers20 were used. The PCR product, with positive and negative controls, was subjected to electrophoresis in a 2% agarose gel stained with ethidium bromide.
DNA extracted from saliva was used for genome analysis of the proband. One microgram of DNA was used, which was labeled and hybridized together with the control DNA (Promega Corporation, Madison, WI) in the NimbleGen Human CGH 12x135K array (Roche NimbleGen, Inc., Reykjavik, Iceland). The array was scanned on a NimbleGen MS 200 Microarray Scanner (Roche NimbleGen, Inc.). Image files from the MS 200 data collection program were imported into DEVA v1.2.1 (Roche NimbleGen Inc.) for analysis. The CGHweb21 program was used, and each genomic region showing a change in copy number was examined by the USCS22 genome browser to determine its location.
Additionally, a bioinformatic analysis was performed with the proteins involved in the genetic alterations found in the patient, together with other genes described in the literature 3, 5, 6, through the STRING23 and CYTOSCAPE24 programs. A correlation was made between the genotype obtained in the bioinformatic analysis and the patient's genotype.
RESULTS
The patient comes from the rural area of the Cotopaxi province. The ancestry study shows the following ethnic composition: Native American 73%, European 23%, and Afro-descendant 4%.
The cytogenetic study in peripheral blood revealed two cell populations, one with 46 chromosomes and sexual formula XX in 74% of metaphases analyzed and another with 46 chromosomes and sexual formula XY in 26% of metaphases. It was checked with the GTG banding technique (Figure 3). The karyotype study of the parents was typical.
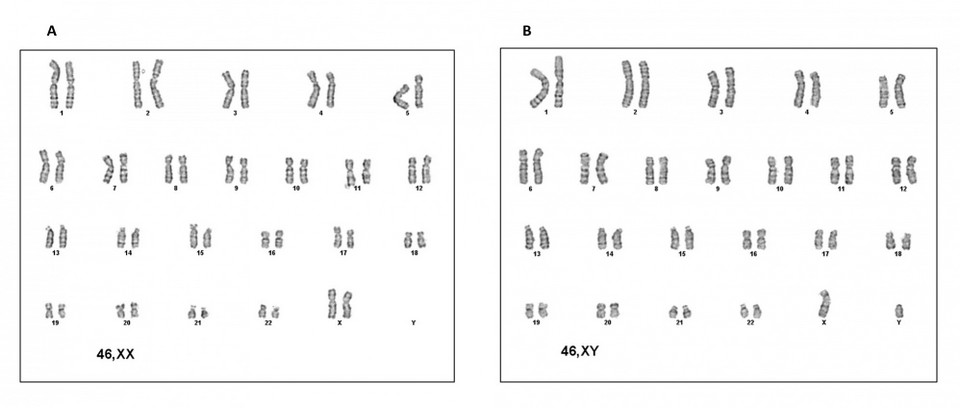
Figure 3. Cytogenetics of the patient. A) XX female karyotype. B) XY male karyotype.
The FISH study confirmed the presence of the two cell lines with signals for the centromere of the XX and XY chromosomes (Figure 4).
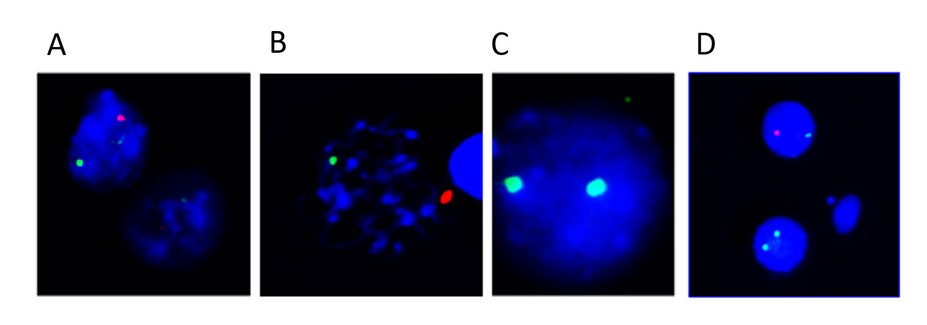
Figure 4. Results of the FISH of the patient. The green signal corresponds to the centromere of the X chromosome and the red signal to the Y chromosome centromere. A) XY. B) XY. C) XX. D) XX and XY
The study of the SRY gene in the patient's peripheral blood was positive (Figure 5).

Figure 5. 2% agarose gel electrophoresis of the PCR product of the SRY gene. Columns 1 correspond to the molecular weight marker, 2 the negative control and 3 the amplified from the patient.
The results of the DNA arrays carried out in another tissue such as buccal mucosa, validated the finding of two cell populations, in addition to evidencing 42,893,491 bp with numerical alterations: 2,230,858 bp in gains and 40,662,633 bp in losses (Figure 6, Table 1).
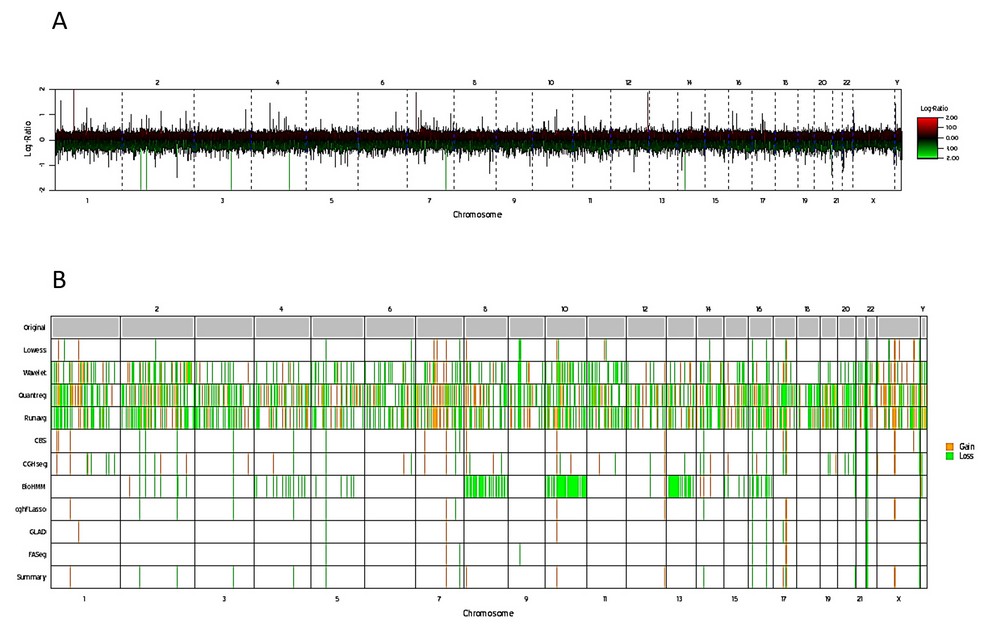
Figure 6. Genetic mapping array of the patient. A) Plot of genome-wide copy number, results are shown for log2 ratio (a measure of chromosome copy number; Y-axis) versus genomic position on each chromosome (red lines up and green lines below) indicate gain and loss respectively; X-axis). B) Whole genome image, gains and losses shown in yellow and green, respectively.
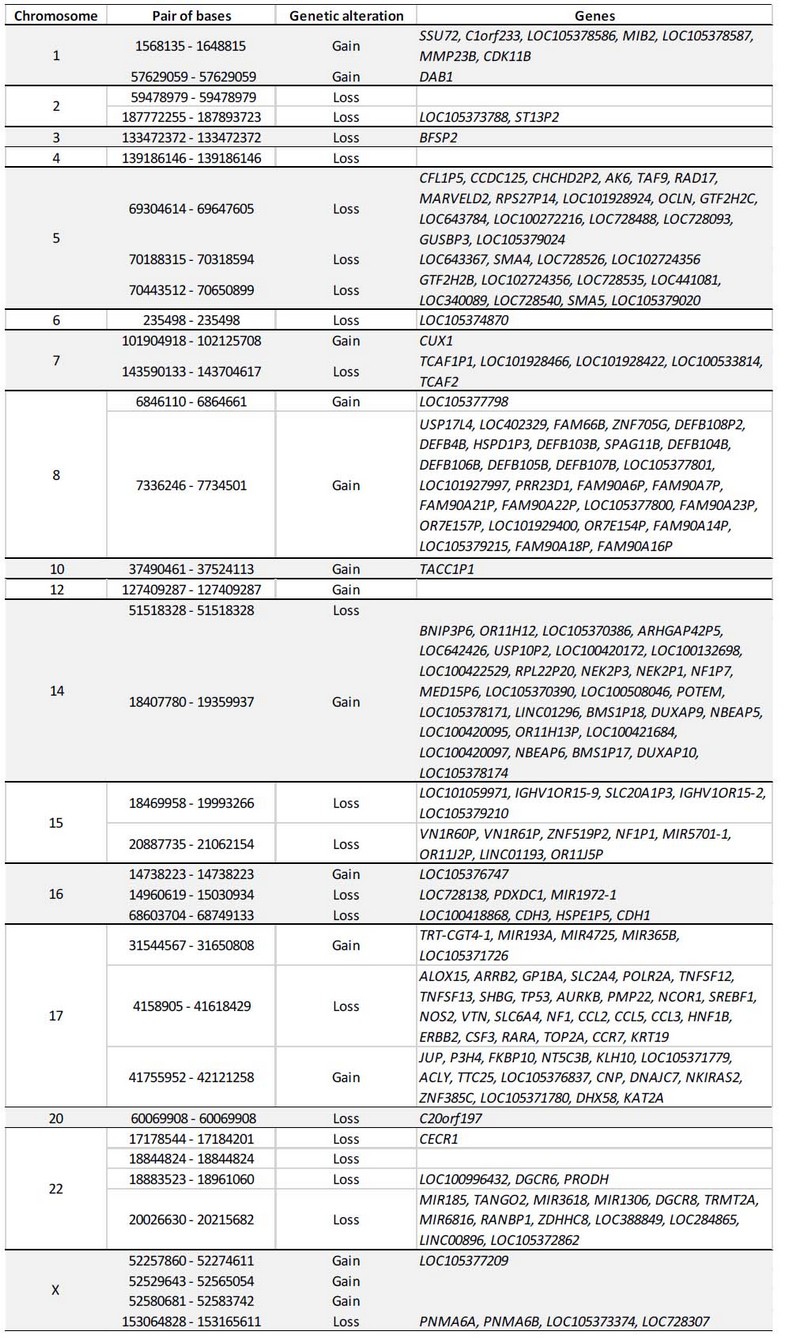
Table 1. Genes involved in the numerical alterations detected by the array.
The protein products of the genes involved in gains and losses in the current patient showed some interactions according to the analysis by STRING (Figure 7). Other genes have individual actions and do not show interactions between genes located in chromosomal regions with numerical alterations:

Figure 7. Interactome of 36 proteins produced by genes located in the regions with numerical alterations by the array. Pink lines, experimentally determined interactions; light blue lines, determined interactions in selected databases; lines of other colors, predicted gene neighborhood interactions, gene fusions, and protein homology.
The analysis of the proteins involved in the genetic alterations found in the patient, together with other genes described in the literature in situs inversus, showed interactions (Figure 8):
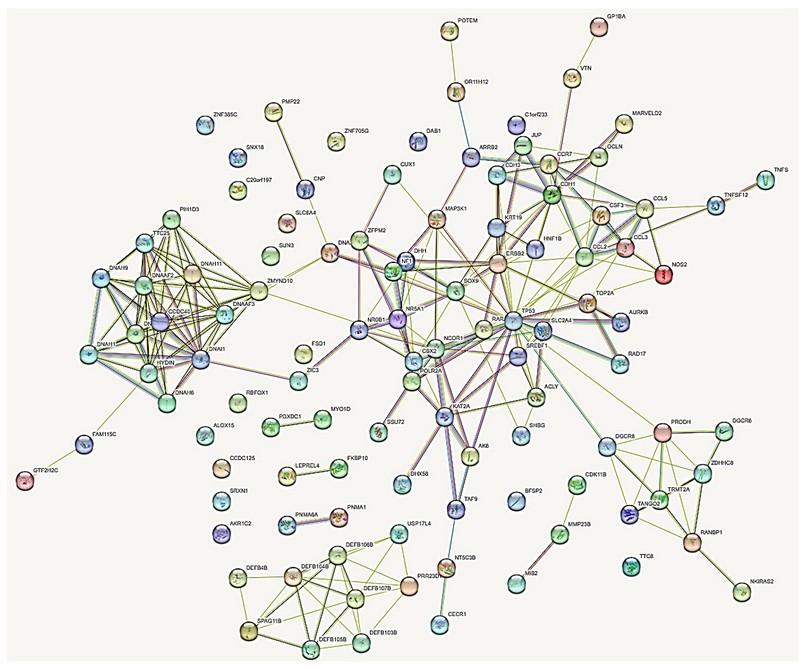
Figure 8. Interactome of 109 protein products of genes located in the regions with numerical alterations by array, and protein products of genes described in the literature. Pink lines, experimentally determined interactions; light blue lines, determined interactions in selected databases; lines of other colors, predicted gene neighborhood interactions, gene fusions, and protein homology.
The protein products of these genes show a relationship according to the interactome with a non-random index of 0.558, which means that these proteins have more interactions with each other than would be expected from a random set of proteins of the same size and degree of distribution extracted from the genome. This association indicates that the proteins are at least partially connected biologically as a group.
In the first in silico filtration of the patient with the proteins reported in the literature, they showed 109 interacting proteins (Additional file 1), with their functions (Additional file 2) and the genotype-phenotype relationship (Additional file 3). Four highly correlated clusters were found with 90 interacting proteins and 16 isolated proteins, and 3 pairs of independently interacting proteins. The interactome analysis evidences the etiopathogenesis of the alterations presented by the proband. Of all these, the following stand out for their function: 5 genes for abnormal gonadal development bipotential to the testis or ovary: CBX2, DHH, NR5A1, SOX9, SRY; 8 genes associated with male sex determination DHH, SOX9, NR0B1, NR5A1, ADA2, RARA, ZFPM2 and SRY; 7 genes involved in the development of primary male sexual characteristics: DHH, NR0B1, NR5A1, RARA, SOX9, SRY and ZFPM2; 5 genes involved in the development of female gonads and steroid hormones: DHH, NR0B1, NR5A1, SREBF1 and ZFPM2; 6 genes of regulatory networks and gene blockers in sex determination: MAP3K1, DHH, NR5A1, SOX9, SRY and CBX2; 4 genes involved in molecular and genetic events in somatic sex determination: DHH, NR5A1, SOX9 and SRY; 7 developmental pathway genes of the different types of testis cells: DHH, NR0B1, NR5A1, RARA, SOX9, SRY and ZFPM2; 1 gene for gonadal function and sexual differentiation involved in the potential role and regulation of sexual development: CBX2; 2 genes involved in the maintenance of gonadal cell fate DGCR6 and NR0B1; 3 genes involved in the development of the adrenal glands: NF1, NR0B1 and NR5A1; 4 genes involved in the ontogeny of fetal Leyding cells: DHH, NR0B1, CDK11B and MMP23B; 3 genes for Sertoli cell differentiation: NR0B1, RAR and SOX9; 2 genes involved in the testicles determination SOX9 and SRY; 2 ovarian infertility genes: NCOR1 and NR5A1; 2 genes involved in the FOXJ1 regulatory network in ciliary associations DAB1 and DHH; 14 genes associated with the dynein axoneme complex necessary for the structure of cilia and lungs: CCDC40, DNAAF2, DNAAF3, DNAAF6, DNAH1, DNAH5, DNAH11, DNAH6, DNAI1, HYDIN, TTC8, ODAD4, ADA2 and ZMYND10; 15 genes associated with ciliary movements and abnormalities: CCD40, DNAAF2, DNAAF3, DNAAF6, DNAA11, DNAH1, DNAH11, DNAH5, DNAH6, DNAH9, DNAI1, HYDIN, TTC8, ZMYND10 and ODAD4; 6 genes that determine left-right asymmetry of the liver, digestive tract, and pancreas: CCDC40, ADNAF2, ADNH5, ZIC3, ADNH11 and ODAD4; 5 positive chemotaxis genes: CCL3, CCL5, DEFB103B, DEFB104B and DEFB4B; 7 bilateral symmetry determining genes: CCD40, ADNAF2, ADNH11, ADNH5, ADNI1, ODAD4 and ZIC3; 2 mammary development genes: CDH1 and TP53; 2 ectoderm development genes: CCL2 and JUP; 2 endoderm development genes: HNF1B and ZIC324.
DISCUSSION
As far as we have reviewed, the association between true hermaphroditism with a XX/XY karyotype resulting from chimerism and situs inversus totalis is exceptional. The present study confirms our approach through the different analyses carried out on the proband.
A plain chest X-ray performs the patient's first sign of visceral transposition. This data, added to subsequent echo sonographic studies, confirmed the diagnosis of situs inversus totalis. Generally, this condition is asymptomatic25, 26.
In the present case, the patient lives as a woman; she presents the secondary sexual characteristics of a woman, and although hypogonadal, she does not present evident anomalies in the external genitalia. On the other hand, the presence of the Y chromosome and the SRY gene explain the hormonal and gonadal data18.
The present patient consulted for hypogonadism; her apparent phenotype is female, with the previously described characteristics. The initial peripheral blood karyotype of the 46, XX/46, XY patient defined her as a true hermaphrodite with a female phenotype, as described in the literature12-15, 18. This led to an exhaustive physical examination and hormonal tests with low normal estradiol and FSH, and abnormally high testosterone, although not normal for a male. This could be explained by the proportion of chimerism XX 74% and XY 26%. Ultrasonography confirmed hypogonadism and visualized apparent bilateral gonads, and the biopsy of the right gonad showed ovotestis, which led to studies of tissues of other embryonic origin16, 17
If testicular tissue is present in the patient's gonad, the fetal Sertoli cells produce the anti-Müllerian hormone, which is responsible for the regression of the Müllerian ducts. At the same time, fetal testicular Leydig cells produce testosterone from cholesterol to generate activated dihydrotestosterone. If the gonad is the ovary, it does not produce the Müllerian-inhibiting substance, and otherwise, the Müllerian ducts would become the uterus, fallopian tubes, and cervix. This may indicate that the patient's androgen secretion was not sufficient to drive scrotal and penile development due to deficiencies in testosterone production, dihydrotestosterone deficiency, androgen insensitivity, and defects in anti-Müllerian hormone or its receptor during lactation embryonic stage15, 17, 18 Our patient has normal female hormones, although low, and present testosterone, although not within normal ranges for a man, but high for a woman—condition explained by the presence of both tissues in the patient, both ovarian and testicular.
Most histopathological studies of ovotestis are mixed ovarian-testicular tissues, as in the case of the current patient. The risk of neoplasia in mixed gonads of the ovotestis type is between 2 and 3%. It is plausible that our patient presents two cell populations derived, according to the literature, from more than one fertilized egg or the union of more than two gametes.16, 27. We do not think our patient is a mosaic, the product of an XXY zygote with subsequent failure in cell division at least two consecutive mitotic events and the formation of two cell lines XX and XY16. Peripheral blood and buccal mucosa studies point to the XX/XY chimeric origin and to a primary chimera, in which the ovum and the second polar body could be fertilized by two spermatozoa that would cause tetra gametic aggregation. We believe that the fertilization theory of the ovum and second polar body is more accurate, which, by uniting in morula phases, would determine the different proportion of the cell populations of our patient, which is also the most frequent way for chimeras to occur10, 12, 16.
One more piece of information that supports hermaphroditism due to a chimera is that congenital adrenal hyperplasia was ruled out since cortisol is normal. It is the most frequent cause of DSD, with which, being its XX/XY karyotype, we reaffirm hermaphroditism true chimeric, additionally confirmed at laparoscopy, which identified ovotestis.
Disorders of Sex Development (DSD) are defined as congenital conditions in which the development of the chromosomal, gonadal or anatomical sex is atypical. DSDs range from mild forms of hypospadias to complete sex reversal. Gonadal anomalies are estimated to be 1 in 4,500 live births17. Hermaphroditism is a rare disorder defined by the presence of testicular and ovarian tissue in the same individual, regardless of the karyotype and has an incidence of 1 in 100,000 live births15.
To this day, the etiology and pathogenesis of true hermaphroditism remain unclear. Still, sex chromosome abnormalities, abnormal gonadal development, and related endocrine disorders during embryonic development and some genes may be involved15, 17. Several studies show that sexual differentiation and gonadal development require the participation of the SRY gene in chromosome Yp11.2 and that this is key for testis development, which in our patient is evidenced by the positive result for the SRY gene and would explain the finding of rudimentary gonads with a positive biopsy for ovotestis. Although other genes have been implicated in DSD, they are not the subject of our study, and we only mention them: the NR0B1 gene on chromosome Xp21.3, the NR5A1 gene on chromosome 9q33, the CBX2 gene on chromosome 17q25, the MAP3K1 gene on chromosome 5q11. 2, DHH gene on chromosome 12q13, AKR1C2 gene on chromosome 10p15, ZFPM2 gene on chromosome 8q23, and SOX9 gene on chromosome 17q2415, 17, 18.
The patient underwent FISH tests in peripheral blood that revealed the XX and XY cell populations. However, to a lesser extent, the latter was validated with the amplification of the SRY gene by PCR.
To check for chimerism, we evaluated DNA from two types of tissues: peripheral blood and buccal mucosa. The results of the karyotype in mesodermal tissue (blood) and of the genetic mapping arrays in ectodermal tissue (mucosa) show two cell populations, one XX and the other XY. This procedure for verifying chimerism has been described in various studies3, 8, 12, 14-16, 28. In our patient, the karyotype is key, since being XX/XY, and having demonstrated that it is a chimera through studies of two tissues, this case is an infrequent finding. We have not found in the literature a case of hermaphroditism of chimeric origin associated with situs inversus totalis.
The etiology of situs inversus totalis is related, among other reasons, to mutations in various genes as reported by OMIM (Online Mendelian Inheritance of Man)29 such as Myosin IC (MYO1D), the genes for ciliary dyskinesia CILD, DNAH1, DNAAF3, DNAI1, genes for visceral heterotaxy (HTX), hairpin box genes (FOXJ), Dynein assembly genes (ADNAF2), genes for the Tetratricopeptide 8 (TTC8) protein, and 50 genes can be included in this list5, 6. Genomic studies of this affectation blame mutations of these genes. Genes with ciliary dyskinesia in 60% of the cases analyzed imply genetic heterogeneity in the disorder's origin, even because many of the individuals analyzed did not present mutations in one or several of the genes6.
Of the chromosomal regions with copy number alterations in the current patient, according to genetic mapping arrays, gains involving the TTC25 gene were found. This gene encodes a protein containing a tetratricopeptide repeat domain located in ciliary axonemes and plays a role in the coupling of the outer arm of dynein to the cilia. Mutations in this gene cause severely reduced ciliary motility and the disorder CILD35 (primary ciliary dyskinesia type 5). Primary ciliary dyskinesia is often associated with recurrent respiratory infections, immobile sperm, and situs inversus, an inversion in left-right body symmetry30.
The interactome analysis ultimately showed the association of genes present in regions with numerical alterations with genes previously described for situs inversus5, 6, including that of TTC25 with DNAI1 also described31. 4 important clusters, 3 medium clusters and 16 isolated proteins were identified, also, at the level of function and correlation with the phenotypic signs of the patient.
Similarly, the in-silico analysis of the interactome of proteins involved in the patient, the corresponding proteins of situs inversus and hermaphroditism, shows a strong genotype-phenotype association.
CONCLUSION
we present a patient with an extremely rare diagnosis in which three pathologies converge: chimerism, hermaphroditism and situs inversus totalis with a strongly associated genotype-phenotype interactome. The combined use of techniques: conventional cytogenetics, FISH, PCR and Arrays allows us to elucidate with better precision the affectation of the patient and the associations between altered genes, the phenotype and the situs inversus. This analysis revives the discussion about using and accessing better technological tools to characterize patients with malformation problems and complex DSDs.
REFERENCES
1. Spoon JM. Situs inversus totalis. Neonatal Netw. 2001; 20(1): 59-63. doi: 10.1891/0730-0832.20.1.63.
2. Casey B. Two rights make a wrong: human left–right malformations. Hum Mol Genet. 1998; 7(10): 1565–1571. doi: 10.1093/hmg/7.10.1565.
3. Mayo CW, Rice RG. Situs inversus totalis. A statistical review of data on seventy-six cases with special reference to disease of the biliary tract. Arch Surg (1920). 1949; 58(5): 724-730.
4. Pennekamp P, Menchen T, Dworniczak B, Hamada H. Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia. 2015; 4(1):1. doi: 10.1186/s13630-014-0010-9.
5. Postema MC, Carrion-Castillo A, Fisher SE, Vingerhoets G, Francks K. The genetics of situs inversus without primary ciliary dyskinesia. Sci Rep. 2020;10(1): 3677. doi: 10.1038/s41598-020-60589-z.
6. Ye Y, Huang Q, Chen L, Liang C, Zhuang K, Liu T, Yuan F, Zhang X, Li J, Dang H, Chen R, Fu Y, Yue Y. Pathogenic variants identified using whole-exome sequencing in Chinese patients with primary ciliary dyskinesia. Res Square. 2021; doi.org/10.21203/rs.3.rs-156279/v2.
7. Yu N, Kruskall MS, Yunis JJ, Knoll JHM, Uhl L, Alosco S, Ohashi M, Clavijo O, Husain Z, Yunis EJ, Yunis JJ, Yunis EJ. Disputed maternity leading to identification of tetragametic chimerism. N Engl J Med. 2002; 346(20): 1545-1552. doi: 10.1056/NEJMoa013452.
8. Quirós-Alpíar JL, Alpízar-Miranda KE. Quimerismo genético un nuevo paradigma para la medicina legal. Med Leg Costa Rica. 2009; 26(2): 73-78.
9. Schoenle E, Schmid W, Schinzel A, Mahler M, Ritter M, Schenker T, Metaxas M, Froesch P, Froesch ER. 46,XX/46,XY Chimerism in a phenotypically normal man. Hum Genet. 1983; 64(1): 86-89. doi: 10.1007/BF00289485.
10. Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB. Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet. 2010; 19(7): 1263-1275. doi: 10.1093/hmg/ddq003.
11. De Carvalho AFL, Pitanga PML, Alves ES, Miguel DSCG, Santo LDE, de Araújo AEF, Ornellas ACP, Toralles MBP. Chimerism 47,XY,+8/46,XX: Follow-up for 11 years. J Pediatr Genet. 2020; 12(1): 81-85. doi: 10.1055/s-0040-1721440.
12. Madan K. Natural human chimeras: A review. Eur J Med Genet. 2020; 63(9): 103971. doi: 10.1016/j.ejmg.2020.103971.
13. Walker AM, Walker JL, Adams S, Shi E, McGlynn M, Verge CF. True hermaphroditism. J. Paediatr Child Health. 2000; 36(1): 69-73. doi: 10.1046/j.1440-1754.2000.00432.x.
14. Modan-Moses D, Litmanovitch T, Rienstein S, Meyerovitch J, Goldman B, Aviram-Goldring A. 2003 True Hermaphroditism with ambiguous genitalia due to a complicated mosaic karyotype: Clinical features, cytogenetic findings, and literature review. Am J Med Genet A. 2003; 116A(3): 300-303. doi: 10.1002/ajmg.a.10869.
15. Chen CQ, Liu Z, Lu YS, Pan M, Huang H. True hermaphroditism with dysgerminoma: A case report. Medicine (Baltimore). 2020; 99(22): e20472. doi: 10.1097/MD.0000000000020472.
16. West JD, Everett CA. Preimplantation chromosomal mosaics, chimaeras and confined placental mosaicism. Reprod Fertil. 2022, 3(2): 66-90. doi: 10.1530/RAF-21-0095.
17. Ohnesorg T, Vilain E, Sinclair AH. The genetics of disorders of sex development in humans. Sex Dev. 2014; 8(5): 262-272. doi: 10.1159/000357956.
18. Josso N, Rey RA. What does AMH tell us in pediatric disorders of sex development? Front Endocrinol (Lausanne). 2020; 11: 619. doi: 10.3389/fendo.2020.00619.
19. Zambrano AK, Gaviria A, Santiago Cobos-Navarrete S, Gruezo C, Rodríguez-Pollit C, Armendáriz-Castillo I, García-Cárdenas JM, Santiago Guerrero S, López-Cortés A, Leone PE, Pérez-Villa A, Guevara-Ramírez P, Yumiceba V, Fiallos G, Vela M, Paz-y-Miño C. The three-hybrid genetic composition of an Ecuadorian population using AIMs-InDels compared with autosomes, mitochondrial DNA and Y chromosome data. Sci Rep. 2019; 9(1): 9247. doi: 10.1038/s41598-019-45723-w.
20. Mendes JR, Strufaldi MW, Delcelo R, Moisés RC, Vieira JG, Kasamatsu TS, Galera MF, Andrade JA, Verreschi IT. Y-chromosome identification by PCR and gonadal histopathology in Turner's syndrome without overt Y-mosaicism. Clin Endocrinol (oxf). 1999; 50(1): 19-26. doi: 10.1046/j.1365-2265.1999.00607.x.
21. Lai W, Choudhary V, Park PJ. CGHweb: a tool for comparing DNA copy number segmentations from multiple algorithms. Bioinformatics. 2008; 24(7): 1014-1015. doi: 10.1093/bioinformatics/btn067.
22. UCSC genome browser on Human 2023. [Internet]. [Consultado Mar 2023]. Disponible en: http://genome.ucsc.edu
23. STRING CONSORTIUM 2023. [Internet]. [Consulted Mar 2023]. Available in: https://string-db.org/cgi/network?pollingId=bffa7HaK3v8v&sessionId=bM432PEbQqLV&urldisam=bbu90dDhE6i8.
24. CYTOSCAPE 2023. [Internet]. [Consulted Mar 2023]. Available in: https://cytoscape.org/
25. Segal, NL. Situs inversus totalis in twins: A brief review and life history / Twin research: Twin studies of Trisomy 21; Monozygotic twin concordance for bilateral coronoid hyperplasia; Prenatal hormonal effects in mixed-sex non-human primate litters; Insurance mandates and twinning after in vitro fertilization / News reports: First report of identical twin puppies; Twins sisters turn 100; Remembering an identical twin production designer; New York city marathon quadruplets. Twin Res Hum Genet. 2017; 20(1):90-95.
26. Gentile BA, Tighe DA. Situs inversus totalis. N Engl J Med. 2019; 380(24): e45. doi:10.1056/NEJMicm1811002.
27. McLaren A. Mammalian chimaeras. 1976; Cambridge University Press, Cambridge https://books.google.com.ec/books?hl=es&lr=&id=ULE8AAAAIAAJ&oi=fnd&pg=PA1&ots=yASFQEN0Xy&sig=N6Rl0dtayWALgqZmNrmM9HoQnrE&redir_esc=y#v=onepage&q&f=false.
28. Nelson JL. Your cells are my cells. Sci Am. 2008; 298(2): 64-71.
29. OMIM, Online Mendelian Inheritance in Man 2023. [Internet]. [Consulted Mar 2023]. Available in: https://www.omim.org/
30. GeneCards 2023. [Internet]. [Consulted Mar 2023]. Available in: https://www.genecards.org/
31. De Ita M, Gaytán-Cervantes J, Cisneros B, Araujo MA, Huicochea-Montiel JC, Cárdenas-Conejo A, Lazo-Cárdenas CC, Ramírez-Portillo CI, Feria-Kaiser C, Peregrino-Bejarano L, Yáñez-Gutiérrez L, González-Torres C, Rosas-Vargas H. Clustering of Genetic Anomalies of Cilia Outer Dynein Arm and Central Apparatus in Patients with Transposition of the Great Arteries. Genes (Basel). 2022; 13(9): 1662. doi: 10.3390/genes13091662.
Received: May 15, 2023/ Accepted: June 10, 2023 / Published: June 15, 2023
Citation: Paz-y-Miño C, García J L, Hernández-Rivas J M, Leone P E. Cytogenetic, genomic and interactome evaluation in an unusual case of a patient with XX/XY chimerism, hermaphroditism and association with situs inversus totalis. Revis Bionatura 2023;8 (2) 62. http://dx.doi.org/10.21931/RB/2023.08.02.62