CS 2019.02.01.19
Files > Conference Series > 2019 > Humboldt Kolleg 2019
Bionatura Conference Series Vol 2. No 1. 2019
“Breaking Paradigms: Towards a Multi-, Inter- and Transdisciplinary Science” In commemoration of the 250th Anniversary of Alexander von Humboldt
Sickle Cell Anemia: A review on the most severe form of Sickle Cell Disease

María Belén Paredes, María Eugenia Sulen
available in: http://dx.doi.org/10.21931/RB/CS/2019.02.01.19
ABSTRACT
Sickle cell disease (SCD) is a group of hereditary disorders caused by a single point mutation in the β-globin gene. This mutation results in the formation of a mutated hemoglobin S (HbS) and the consequent sickle phenotype of erythrocytes. SCD is common in regions of malaria endemicity. However, changes in population dynamics enabled the movement of the mutated gene to other areas such as North America and Europe. Sickle cell anemia (SCA) is the most severe form of SCD and affects millions of people around the globe. The clinical manifestations of SCA arise primarily from the polymerization of deoxygenated hemoglobin S (deoxyHbS) leading to vascular occlusion and hemolytic anemia. Clinical complications of the disease are derived from deoxyHbS polymerization, but there are several therapeutic strategies to reduce the severity of the symptoms. Gene therapy has arisen as a new therapeutic approach aimed to cure rather than to treat the symptomatology of SCA by targeting the altered β-globin gene for gene correction.
INTRODUCTION
Sickle cell disease (SCD) refers not to a single disease but to a group of inherited hematopoietic disorders that are considered one of the most common genetic disorders. SCD is an increasing global public health disorder that affects globally millions of people and is related to significant morbidity and mortality. They are monogenic disorders caused by a point mutation in the β-globin gene (HBB) which polymerizes an abnormal version of hemoglobin upon deoxygenation. SCD includes several inherited hemoglobinopathies with variable phenotypes in which the heterogeneity appears from different intrinsic characteristics of the sickle erythrocytes. SCD hemoglobinopathies divide into two groups, structural variants that produce abnormal hemoglobin and thalassaemias that have a lack or decreased synthesis of the globin chains. They commonly include sickle cell anemia (homozygous HbSS), HbS β-Thalassaemia, HbSC, HbSD, and HbSE. The anomalous version of hemoglobin (HbS) results in a dramatic physical deformation of erythrocytes, giving them a sickle shape and altered surface properties. As a consequence of the sickle shape of erythrocytes, SCD hemoglobinopathies exhibit vaso-occlusion, hemolysis, several degrees of anemia and endothelial dysfunction, which result in acute and chronic pain in muscles and bones, damage in the most vital organs such as brain, kidneys, liver, spleen and lungs, and increased susceptibility to infections. The most common type of SCD is sickle cell anemia (SCA, which is generally confused with sickle cell disease; however, they are not the same, and the terms cannot be used interchangeably. This study will focus on Sickle cell Anemia as it is the most severe type of SCD.1-8
Epidemiology
While sickle cell anemia (HbSS) results from the inheritance of the HbS alleles from both parents (homozygosity for the HbS allele), the sickle cell trait is due to heterozygosity for the HbS. Individuals with the sickle cell trait are carriers of the HbS allele, produce both normal (HbA) and abnormal hemoglobin (HbS), and do not present the severe clinical symptoms of SCD. Instead, they present a survival advantage over both HbSS and HbAA homozygotes against falciparum malaria infections. The distribution of the HbS gene in Africa reflects a very close association with malaria endemicity, with the higher frequency of HbS found in tropical areas of low altitude and with substantial amounts of annual rainfall. SCD is thought to have arisen from spontaneous mutations in the HBB gene across different regions. The description of several haplotypes found on the chromosome 11th namely, Bantu, Benin, Cameroon, Senegal, and Arab– Indian, corresponding to the geographical areas with a high prevalence of the disease, support this hypothesis.1,4,7,9,10
In populations where malaria is present the prevalence of hemoglobin disorders ranges from 0.3 to 25 per 1000 live births as SCD provides a relative resistance against falciparum malaria for which the specific role of the mechanism is still being debated. The sickle cell gene is predominant in Sub-Saharan Africa, the Caribbean, Asia, Mediterranean, and southern Europe and the Middle East due to the exceptional level of protection that it provides against malaria to carriers of the HbS allele, which makes them less likely to get the disease and run parasite counts. The most severe forms of SCD are found in Sub-Saharan Africa and the mildest form in the Indian-Arab haplotype. Also, almost all white patients with SCD are of Mediterranean origin. The transatlantic slave trade, massive migration, and changes in population dynamics are responsible for the movement of the mutated gene to geographic regions where malaria is not endemic such as North America and the United Kingdom, thus, impacting the epidemiology of SCA.1,7,8,11,12
Classification of SCD
The group of SCD blood disorders includes hemoglobinopathies in which the sickle HBB gene is inherited at least from one parent. The structural variants of SCD include sickle cell anemia (SCA) which is caused by the homozygous sickle HBB gene, which is inherited from both parents. It is caused by a point mutation that replaces glutamic acid with valine in the sixth amino acid position A-to-T in the first exon of the HBB gene. Other variants that comprise SCD emerge from the co-inheritance of the sickle HBB gene with another abnormal HBB gene. These types of co-inherited SCD include HbSC, which results from the coinheritance of the sickle HBB gene with the HbC gene in which glutamic acid is replaced with lysine in the sixth amino acid position. HbSC represents globally one-third of the total SCD cases. Another co-inherited SCD is HbSD which is a rare type of β-globin variant in which the glutamic acid is replaced with glycine at codon 121. HbSE is another rare variant of SCD in which there is a change from glutamic acid to lysine in the twenty-sixth amino acid position. SCD also includes β-thalassaemias, which can be caused by more than 200 different mutations and in rare cases by deletions. The β-thalassaemias can be either caused by absent synthesis of β-globin chains (0-thalassemia) or decreased synthesis of β-globin chains (+-thalassemia).11-19
Genetics and disease mechanism for SCA
Sickle cell disease is a multisystem, monogenic, autosomal recessive disorder. Sickle cell anemia is the most common and most severe genotype of sickle cell disease, and it was the first disease for which the molecular mechanism was described. This disease is caused by the presence of a mutated form of hemoglobin A known as hemoglobin S (HbS) in red blood cells. Hemoglobin is a tetrameric molecule composed of two heterodimers. Fetal hemoglobin (HbF) has two alpha chains and two gamma chains (α2γ2), whereas adult hemoglobin A (HbA) has two alpha and two beta chains (α2β2). HbF is the predominant hemoglobin during the most part of gestation and the first two months after birth. After that, adult hemoglobin A predominates due to a switch in the expression of the gene encoding for the γ-globin (HBG) to the expression of HBB which codes for the β-globin polypeptide chain in erythroid progenitors. This change in gene expression results in the synthesis of HbA instead of HbF. In individuals with sickle cell anemia, there is a delayed switch in the expression from HBG to HBB and the subsequent replacement of HbF by HbS. HbS shortens the life span of red blood cells from ~120 days when HbA is present to ~16 days.4,6,8,9,20-22
Sickle cell anemia is caused by an A-to-T point mutation in the 17th nucleotide of the first exon of the human β-globin gene (HBB) which is located in the short arm of chromosome 11 at position 15.4 (11p15.4). This nucleotide switch results in glutamic acid being substituted by valine in the sixth position on the two β-subunits of hemoglobin A (HbA), leading to the formation of an abnormal hemoglobin S tetramer, (α2βS2 HbS) with altered net charge and conformation. Under oxygen deprivation, the missense mutation generates a hydrophobic motif in the deoxygenated HbS tetramer (deoxyHbS) which causes the binding between β1 and β2 chains of two hemoglobin molecules. The polymerization distorts the architecture of red blood cells resulting in their physical deformation and alteration of rheological features, including diminished flexibility. HbS polymerization depends on the concentration of deoxyHbS, intracellular HbS concentration, and the presence of fetal hemoglobin.2,6-9,23,24
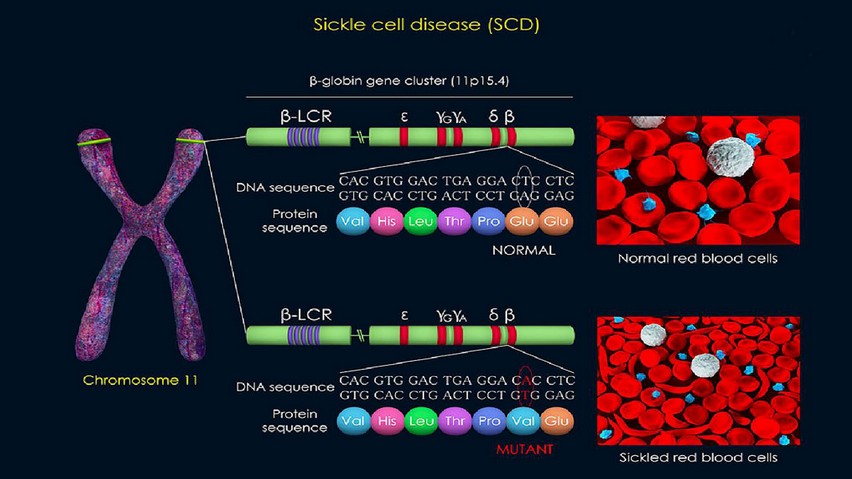
Figure 1. 3d render of the beta-globin mutation that leads to sickle cell disease. ID 115531659 © Meletios Verras | Dreamstime.com
Pathophysiology of SCA
The polymerization of deoxyHbS is the basis of the pathophysiology of SCA, and it is the primary determinant of disease severity. Polymerization is promoted by both hyperosmolarity, which increases the concentration of hemoglobin in RBCs, and acidosis, which decreases the affinity of HbS for oxygen. The sickle shape of red blood cells resulting from the polymerization of deoxyHbS promotes intravascular hemolysis and vaso-occlusion, the two major distinctive features of sickle cell anemia. Hemolysis arises from damaged sickle RBC membranes and leads to chronic anemia. Because the lifespan of sickle RBCs is shorter than the lifespan of normal erythrocytes, sickle RBCs are prematurely lysed. Damaged RBCs are removed from circulation by extravascular and intravascular hemolysis. During the latter, the release of Hb into the circulation generates oxidative stress, depletes nitric oxide, and releases heme groups promoting inflammation and causing vascular injury. Elevated plasma heme levels increase expression of cell adhesion molecules (CAMs) on endothelial cells, which promotes the recruitment of neutrophils and its subsequent interaction with erythrocytes and other cell types. To compensate for the reduced number of RBCs, the bone marrow increases erythropoiesis. However, this compensation is incomplete, resulting in chronic anemia, which exerts additional stress on the cardiovascular system. As a result of increased erythropoiesis, the bone marrow hypertrophies and stress reticulocytes are produced, which are the first cells that adhere to the endothelium, enabling vaso-occlusion.2,3,20,23,25-27
Vaso-occlusion is the result of the adhesion of sickle RBCs and leukocytes to the vascular endothelium leading to reduced blood flow, tissue ischemia, and organ damage. Vaso-occlusion is triggered by the inflammatory stimuli from hemolysis that activates endothelial cells, which in turn leads to the recruitment of circulating neutrophils to postcapillary venules. P-selectin, a cell adhesion molecule that is expressed in activated endothelial cells, initiates the adhesion of recruited leukocytes to the endothelium and mediates abnormal rolling and adhesion of sickle erythrocytes to blood vessels. Formation of aggregates between sickle RBCs and neutrophils and neutrophils and activated platelets is mediated by activated Mac-1 (macrophage-1 antigen) expressed on adherent neutrophils and P-selectin, respectively. The formation of cellular aggregates results in the obstruction of blood flow. Vascular stasis induces ischemia, thus enhancing RBC sickling and promoting further recruitment of leukocytes. Microvascular occlusion and ischemia are followed by restoration of blood flow, which further promotes tissue injury mediated by reperfusion. Cycles of ischemia and reperfusion cause both oxidant and inflammatory stress leading to activation of vascular oxidases increased expression of endothelial CAMs, increased the synthesis of inflammatory cytokines, and leukocytosis.3,9,23,28
Hemolytic anemia and vaso-occlusion trigger a cascade of events such as vascular-endothelial dysfunction, vasculopathy, hypercoagulation, oxidative stress, nitric oxide dysfunction, and inflammation. This chain-reaction of pathological events leads to a broad range of life-threatening complications by producing irreversible multiorgan damage.4,6
Clinical Complications of SCA
SCD complications are known to arise from intravascular hemolysis and vaso-occlusion. However, the exact mechanisms contributing to the complex manifestation of such pathological events remain to be elucidated. Clinical complications can be broadly classified into acute and chronic complications. The latter can be divided into those related with large-vessel vasculopathy and those that are the product of progressive ischemic organ damage. Acute complications such as acute pain crisis (APC), acute chest syndrome (ACS), and stroke are typically the result of vaso-occlusion. Acute pain crises are the hallmark of SCD and reflect vascular occlusion, compromised oxygen supply, and infarction-reperfusion injury. APCs are responsible for 90% of hospitalizations in the United States and the United Kingdom, with 30% of patients reporting daily symptoms. Recurrent and chronic pain is typically associated with sleep disturbances, anxiety, and depression, this, together with the excessive burden of the disease, results in a poor quality of life. ACS is the most common example or organ failure in patients with SCA and one of the main causes of hospitalization and death. It is the result of vascular occlusion in the lungs, and it includes components from infection, infarction, fat embolism, and pulmonary sequestration. ACS is characterized by chest pain, cough, and dyspnea. Increase in the incidence of both acute painful episodes and acute chest syndrome has been associated with leukocytosis, low HbF levels, and higher hemoglobin plasma concentrations. Stroke affects 10% of SCD patients. Silent strokes are the most common cerebrovascular accident in children with SCD and are the cause of irreversible brain damage with cognitive, physical, and psychological impairments. In adults, strokes are predominantly hemorrhagic, which have a poor prognosis.1,4,5,11,23
Chronic complications associated with vasculopathy, including pulmonary hypertension (PH), chronic leg ulcers, and priapism are the result of low steady-state hemoglobin plasma levels and hyperhemolysis. PH prevalence ranges from 6% to 13% in SCD patients, and it is associated with increased mortality. Chronic leg ulcers affect 8% of SCA patients. Patients suffering from these non-healing wounds show an increased likelihood of having severe hemolysis, making them prone to develop other complications such as PH and priapism. Inflammation and hemolytic anemia are associated with priapism. It can occur at any age but has its peak in the third decade. Both priapism and chronic leg ulcers are debilitating complications of SCD that reduce the quality of life of patients. Chronic complications associated with progressive ischemic organ damage include renal failure, hyposplenism, and avascular necrosis (AVN). Renal failure is another cause of mortality and occurs in 5% to 18% of SCD patients. Kidney disease is thought to be the result of hemolysis; it affects glomerular and tubular function leading to proteinuria and albuminuria. The estimated glomerular filtration rate (eGFR) in patients with SCD decreases with age. Because low eGFR appears concomitantly with high systolic blood pressure and high tricuspid regurgitant velocity (TRV), it has been suggested that chronic kidney disease may be associated with cardiovascular complications. Hyposplenism is one of the significant causes of illness and death in infants because it renders the children susceptible to septicemia and splenic sequestration. Finally, AVN of the femoral head has been reported in 30% of SCD patients. It results from reduced blood supply to the head of the femur which leads to pain and eventual collapse of the hip joint.3-5,11,12,23,29,30
Diagnosis for SCA
The most reliable methods for SCD diagnosis are electrophoretic techniques such as isoelectric focusing (IEF), capillary electrophoresis (CE), and high-performance liquid chromatography (HPLC). These techniques are based on the separation of hemoglobin molecules by differences in isoelectric point, migration velocity, and protein-column interaction times; and allow the differentiation between sickle-cell trait and SCA samples. Point of care diagnostics includes the SickleDex test that differentiates SCA and SCT from normal blood based on solubility differences in HbA and HbS molecules. This test, however, cannot distinguish SCA from SCT. Furthermore, SickleDex, as other chemical and solubility tests, is prone to error, and it should be used with other diagnostic tools to give an accurate diagnosis. Lateral flow assays such as SickleSCAN and HemoTypeSC rely on monoclonal or polyclonal antibodies specific to HbA, HbS, HbF, and HbC. These immunoassays can accurately detect the presence of different hemoglobin variants and can discriminate SCA from SCT and healthy blood.30,31
Early diagnosis is essential to reduce the risk of death from life-threatening SCA complications. Newborn screening in developed countries has allowed early intervention and treatment of SCA, resulting in the reduction of childhood mortality and improvement in the quality of life of adults. On the other hand, low-income countries of sub-Saharan Africa lack robust SCA newborn screening programs. Even though low-cost therapies such as hydroxyurea treatment and prophylaxis are available in these regions, the lack of appropriate diagnostic tools results in 50% to 80% of children dying from SCA complications before the age of 5. Although the diagnostic methods have high sensitivity and specificity, most of them require expensive equipment and extensive training. To reduce SCA-related mortality in developing countries, the development of simple, rapid, and inexpensive screening tests is required.31,32
THERAPEUTIC STRATEGIES TO TREAT SICKLE CELL ANEMIA
Transfusion
SCA is characterized by a hypercoagulable state, which increases the risk for thromboembolic complications, including ischemic stroke, which can cause severe neurocognitive and motor sequelae. Red blood cell (RBC) transfusion is among the therapeutic strategies for treating Sickle cell anemia due to the capability to prevent the development of stroke in patients with elevated transcranial Doppler (TCD), reduce the HbS concentration to prevent sickle-cell associated complications, treat acute chest syndrome and reduce vaso-occlusion related pain. However, RBC transfusion has critical side effects such as transfusion reactions, iron overload, and alloimmunization.33-35
Hydroxyurea (Hydroxycarbamide)
A pharmacologic strategy to treat SCA is using hydroxyurea a myelosuppressive agent, which can induce HbF through altered erythroid kinetics and soluble guanylyl cyclase activation, inhibiting the polymerization of HbS, therefore preventing the sickling of RBCs. Besides, hydroxyurea promotes lower reticulocyte and neutrophil counts marrow cytotoxicity and inhibition of ribonucleotide inhibition. It also reduces hemolysis through improved macrocytosis, intracellular sickling, and erythrocyte hydration. It stimulates improved rheology of circulating reticulocytes and neutrophils and decreased adhesiveness. Hydroxyurea also promotes nitric oxide (NO) release with a possible improved vascular response and local vasodilation. Possibly the most relevant known mechanism of action of hydroxyurea is the inhibition of ribonucleotide reductase (RR) leading to a reduction in intracellular deoxynucleotide triphosphate pools, DNA synthesis, and eventual cell cytotoxicity. Despite the benefits of hydroxyurea, there are still some concerns regarding its safety due to it's short- and long-term toxicity profile.36,37
Hematopoietic Stem Cell (HSC) Transplantation
Allogenic HSCT can be used as vectors to replant genes that are essential for normal hematopoiesis either as bone marrow (BM) or umbilical cord blood (UCB) as stem cell source from matched donors. The major obstacle in HSC transplantation is that it is restricted to donor availability and the probability of finding a matched donor. It is recommended that patients with SCA are transplanted as early as possible.38,39
Iron Chelation
As mentioned previously, RBC transfusions despite preventing stroke they have significant side effects that include iron overload. This is because the human body cannot excrete excess iron. Transfused RBCs at the end of their lifespan are phagocytosed by reticuloendothelial macrophages in the spleen, liver and bone marrow. In this process, at first the iron is stored in the reticuloendothelial macrophages, and the hemoglobin is digested. However, when the capacity of the macrophages is limited, the iron is released to the plasma where it binds to transferrin. When iron concentration increases, transferrin saturation also increases, leading to the recruit of hepatocytes to aid in storage of excess iron. When hepatocytes can no longer store more iron, non–transferrin-bound iron appears in the plasma and enters cardiomyocytes, hepatocytes, pancreatic beta cells and pituitary cells leading to the generation of reactive oxygen species that result in damage to DNA, proteins, lipids, and organelles and eventually cell dysfunction necrosis and apoptosis. In SCA iron-induced complications can result in pancreatic and cardiac iron deposition and liver disease with cirrhosis. For this reason, a therapy with chelating agents that can form complexes with iron and stimulate its excretion to preserve safe iron levels in the body. The iron-chelating agents that can be used include parenteral deferoxamine mesylate, oral deferasirox, and synthetic deferiprone.40
Other therapeutic strategies
Other attractive therapeutic approaches for treating SCA include drugs that target cell adhesion since adhesive interactions assist in vaso-occlusion; for this reason, drugs that target selectin-mediated adhesion are studied. Heparins can inhibit P-selectin adhesive interactions; for this, derivatives of low molecular weight heparin such as Sevuparin are studies as P-selectin blockers. Beta blockers can also be used to target signaling pathways that activate adhesion molecules to reduce RBC adhesion. Also, poloxamer is a nonspecific inhibitor of adhesion that can lower blood viscosity and decrease friction along the vessel wall. Another approach to treat SCA is by targeting inflammatory pathways as platelets, leukocytes, and several proinflammatory pathways contribute to SCA. These inflammatory pathways can be targeted using adenosine. Invariant natural killer (NK) T cells are involved in ischemia. These cells can be downregulated by the activation of the adenosine A2A receptor. Other inflammatory mediators that can be targeted are leukotrienes which are involved in the pain of SCA or inhibiting leukotriene synthetic enzyme 5-lipoxygenase (5-LO) to interfere with leukotriene formation. Also, inflammation can be reduced via inhibition of neutrophil adhesion using intravenous g-globulin (IVIg). Another approach for treating SCA is to use HbF inducing drugs such as decitabine and tetrahydrouridine, arginine butyrate, and pomalidomide in addition to hydroxyurea. Finally, another method is the use hemoglobin anti-sickling and modification agents such as carbon monoxide which can reverse or prevent HbS polymerization, Aes-103 which shifts the oxyhemoglobin dissociation curve to the left, senicapoc which prevents cell dehydration or sanguinate which reduces sickling ad increases Hb levels.41
Gene therapy Strategies to treat SCA
Most therapeutic strategies to treat SCA are targeted to reduce the symptoms of the disease rather than to cure the disease itself. The only currently available cure is the allogeneic HSC transplant. However, the application of this therapy is highly restricted to donor availability and donor-host compatibility. Thus, there has been an increasing interest in the development of autologous gene therapy techniques to correct the patient’s HSCs, so they produce healthy RBCs with wild-type hemoglobin. To achieve this objective, research has been undertaken to achieve efficient and safe gene transfer using viral vectors. Although the first attempts were focused on developing integrating gammaretroviral vectors for β-globin gene transfer, later was found that lentiviral vectors offer some advantages over gammaretroviral vectors. For instance, insertional oncogenesis has not been reported for lentiviral vectors. Also, lentiviruses have the tendency to insert in transcriptionally active regions rather than in transcriptional start sites as in the case of gammaretroviruses, thus, decreasing the potential of altering gene expression. Furthermore, lentiviral vectors can transduce nondividing HSCs and have a safer integration profile because most of them have a self-inactivating design.6,42,43
Other novel gene therapy strategies for site-specific gene correction include the use of targeted nucleases such as CRISPR-associated Cas9 nuclease, homing endonucleases, zinc-finger nucleases (ZFNs), and transcription-activator-like effector nucleases (TALENs), which can create site-specific double-strand breaks in DNA. These therapeutic options rely on the endogenous eukaryotic DNA-repair mechanisms: nonhomologous end joining (NHEJ) and homology-directed repair (HDR). Of the two, HDR is the most attractive mechanism for gene correction because NHEJ repair can lead to errors at the break site, thus, increasing the possibility of disturbing β-globin production by knocking out gene function.42-44
ZFNs have been used to specifically target the β-globin gene and induce high levels of DNA cleavage in human CD34+ hematopoietic stem and progenitor cells (HSPCs). β-globin–targeted ZFNs were delivered with a repair template via an integrase-defective lentiviral vector (IDVL) or with an oligonucleotide donor and affected efficient gene correction via homology-directed repair at the β-globin locus in HSPCs. It was shown that modified HSPCs were able to differentiate into erythroid and myeloid lineage both in vivo in immunodeficient mice and in vitro. Also, gene correction of bone marrow CD34+ cells from patients with SCD led to the production of wild-type hemoglobin (HbA) in cell culture. Despite successful transplantation of modified human cells into immunodeficient mice, the levels of gene correction decreased in the spleen and bone marrow after about four months following engraftment. This suggests that gene correction is more efficient in downstream progenitors than in more primitive HSCs, which are the ultimate target to achieve favorable and continuous clinical effects.42
A self-inactivating lentiviral vector was used to transfer a modified HBB encoding an antisickling variant of the β-globin gene (βA-T87Q) to a patient homozygous for HbS. CD34+ HSCs were obtained from the patient’s bone marrow and were transduced with the LentiGlobin BB305 vector. Following engraftment of transduced HSCs, normal blood cell counts were reached in all lineages, suggesting that the modified HSCs were able to differentiate into lymphoid and myeloid cells. Also, a high concentration of therapeutic HbAT87Q was achieved. It was suggested that the expression of HbAT87Q could suppress intravascular hemolysis because stable hemoglobin concentrations were attained following gene addition. Furthermore, an overall improvement in SCA-associated clinical events and SCA markers was observed. A similar study using the LentiGlobin BB305 Drug Product was carried out in a subject with SCA. The patient produced 51.5% of antisickling Hb with 48% of HbAT87Q, and SCD-related events were not further seen in the patient even after stopping the transfusion therapy.6,45
Because it has been noted that SCD patients with high levels of HbF have extended survival and mild SCD clinical manifestations, other genome-editing techniques are based on increasing the production of fetal hemoglobin rather than repairing the SCD mutation. This can be achieved by gene addition of γ-globin or β/γ-globin hybrid. Also, the modification of the β-globin locus has been proposed to mimic the hereditary persistence of fetal hemoglobin (HPFH) by creating small deletions or point mutations in the γ-globin gene promoters. Knocking out the BCL11A repressor of γ-globin by disrupting its coding sequence or by targeting the BCL11A enhancer could also be a potential strategy to treat SCD. However, the role of BCL11A in B-cell development and HSC function have limited the application of this strategy. The transcription factor LRF/ZBTB7A (Pokemon) is also a major repressor of γ-globin. LRF and LRF/ BCL11A knockout erythroid cell line resulted in upregulation of γ-globin. But, similar to BCL11A, the role of LRF in the hematopoietic system has reduced its applicability for SCD treatment.43,44

Table 1. Summary of Gene Therapy Approaches
CONCLUSION
Sickle cell disease is a group of monogenic inherited diseases caused by point mutations in the β-globin gene (HBB) which polymerizes an abnormal version of hemoglobin upon deoxygenation. These altered versions of hemoglobin (HbS) result in dramatic physical deformation of erythrocytes, giving them a sickle shape and altered surface properties. The most common type of SCD is sickle cell anemia (SCA), which is caused by an A-to-T point mutation in the 17th nucleotide of the first exon of the human β-globin gene (HBB). This nucleotide switch results in glutamic acid being substituted by valine in the sixth position on the two β-subunits of hemoglobin A (HbA). The therapeutic strategies to treat sickle cell anemia include red blood cell (RBC) transfusion to reduce the HbS concentration to prevent sickle-cell associated complications. Another treatment uses hydroxyurea to induce HbF through altered erythroid kinetics and soluble guanylyl cyclase activation, inhibiting the polymerization of HbS, therefore avoiding the sickling of RBCs. Hematopoietic stem cell (HSC) transplantation is an approach that allows the replanting of genes that are essential for normal hematopoiesis, and other therapeutic strategies that serve to treat the symptoms of SCA. However, these approaches, in the most part, do not cure the disease they only manage it. For this reason, gene therapy strategies have been investigated to correct the patient’s HSCs, so they produce normal RBCs with wild-type hemoglobin, either using lentiviral vectors or site-specific gene correction with CRISPR-associated Cas9 nuclease, homing endonucleases, zinc-finger nucleases (ZFNs), and transcription-activator-like effector nucleases (TALENs).
REFERENCES
- Anie KA, Green J. Psychological therapies for sickle cell disease and pain. Cochrane Database of Systematic Reviews 2015. doi:10.1002/14651858.cd001916.pub3.
- Pace BS, Ofori-Acquah SF, Peterson KR. Sickle Cell Disease: Genetics, Cellular, and Molecular Mechanisms, and Therapies. Anemia 2012;2012:1–2. doi:10.1155/2012/143594.
- Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016;127:801–9. doi:10.1182/blood-2015-09-618538.
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. New England Journal of Medicine 2017;376:1561–73. doi:10.1056/nejmra1510865.
- Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. Journal of Clinical Investigation 2017;127:750–60. doi:10.1172/jci89741.
- Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene Therapy in a Patient with Sickle Cell Disease. New England Journal of Medicine 2017;376:848–55. doi:10.1056/nejmoa1609677.
- Aneke J, Okocha C. Sickle cell disease genetic counseling and testing: A review. Archives of Medicine and Health Sciences 2016;4:50. doi:10.4103/2321-4848.183342.
- Modell B. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 2008;2008:480–7. doi:10.2471/blt.06.036673.
- Jarrett K, Williams M, Horn S, Radford D, Wyss JM. “Sickle cell anemia: tracking down a mutation”: an interactive learning laboratory that communicates basic principles of genetics and cellular biology. Advances in Physiology Education 2016;40:110–5. doi:10.1152/advan.00143.2015.
- Allison AC. Genetic control of resistance to human malaria. Current Opinion in Immunology 2009;21:499–505. doi:10.1016/j.coi.2009.04.001.
- Ezekekwu CA, Kotila TR, Akingbola TS, Lettre G, Gordeuk VR, Cooper RS, et al. Sickle Cell Disease Clinical Trials and Phenotypes. Journal of Tropical Diseases 2018;06. doi:10.4172/2329-891x.1000259.
- Serjeant GR. The Natural History of Sickle Cell Disease. Cold Spring Harbor Perspectives in Medicine 2013;3. doi:10.1101/cshperspect.a011783.
- Ballas SK. Sickle cell disease: Classification of clinical complications and approaches to preventive and therapeutic management. Clinical Hemorheology and Microcirculation 2018;68:105–28. doi:10.3233/ch-189002.
- Hannemann A, Weiss E, Rees DC, Dalibalta S, Ellory JC, Gibson JS. The Properties of Red Blood Cells from Patients Heterozygous for HbS and HbC (HbSC Genotype). Anemia 2011;2011:1–8. doi:10.1155/2011/248527.
- Colah R, Mukherjee M, Surve R, Gangakhedkar R, Mohanty D. Hemoglobin sickle D Punjab-a case report. Indian Journal of Human Genetics 2005;11:154. doi:10.4103/0971-6866.19536.
- Hardy MJ, Ragbeer MS. Homozygous Hbe and Hbse Disease in a Saudi Family. Hemoglobin 1985;9:47–52. doi:10.3109/03630268508996981.
- Ellory JC. Haemoglobin C Promotes Distinct Membrane Properties in Heterozygous HbSC red Cells. EBioMedicine 2015;2:1577. doi:10.1016/j.ebiom.2015.10.026.
- Rund D, Rachmilewitz E. β-Thalassemia. New England Journal of Medicine 2005;353:1135–46. doi:10.1056/nejmra050436.
- Cao A, Galanello R. Beta-thalassemia. Genetics in Medicine 2010;12:61–76. doi:10.1097/gim.0b013e3181cd68ed.
- Akinsheye I, Alsultan A, Solovieff N, Ngo D, Baldwin CT, Sebastiani P, et al. Fetal hemoglobin in sickle cell anemia. Blood 2011;118:19–27. doi:10.1182/blood-2011-03-325258.
- Wilber A, Nienhuis AW, Persons DA. Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood 2011;117:3945–53. doi:10.1182/blood-2010-11-316893.
- Sankaran VG, Orkin SH. The Switch from Fetal to Adult Hemoglobin. Cold Spring Harbor Perspectives in Medicine 2012;3. doi:10.1101/cshperspect.a011643.
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. The Lancet 2010;376:2018–31. doi:10.1016/s0140-6736(10)61029-x.
- HBB gene - Genetics Home Reference - NIH. U.S. National Library of Medicine.
- Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nature Reviews Nephrology 2015;11:161–71. doi:10.1038/nrneph.2015.8.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of Sickle Cell Disease. Annual Review of Pathology: Mechanisms of Disease 2019;14:263–92. doi:10.1146/annurev-pathmechdis-012418-012838.
- Steinberg MH. Overview of Sickle Cell Anemia Pathophysiology. Sickle Cell Anemia 2016:49–73. doi:10.1007/978-3-319-06713-1_3.
- Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. New England Journal of Medicine 2017;376:429–39. doi:10.1056/nejmoa1611770.
- Benenson I, Porter S. Sickle Cell Disease. Orthopaedic Nursing 2018;37:221–7. doi:10.1097/nor.0000000000000464.
- Ware RE, Montalembert MD, Tshilolo L, Abboud MR. Sickle cell disease. The Lancet 2017;390:311–23. doi:10.1016/s0140-6736(17)30193-9.
- Bond M, Hunt B, Flynn B, Huhtinen P, Ware R, Richards-Kortum R. Towards a point-of-care strip test to diagnose sickle cell anemia. Plos One 2017;12. doi:10.1371/journal.pone.0177732.
- Piety NZ, Yang X, Kanter J, Vignes SM, George A, Shevkoplyas SS. Validation of a Low-Cost Paper-Based Screening Test for Sickle Cell Anemia. Plos One 2016;11. doi:10.1371/journal.pone.0144901.
- Zétola VF. Role of TCD in sickle cell disease: A review. Perspectives in Medicine 2012;1:265–8. doi:10.1016/j.permed.2012.03.016.
- Beverung LM, Strouse JJ, Hulbert ML, Neville K, Liem RI, Inusa B, et al. Health-related quality of life in children with sickle cell anemia: Impact of blood transfusion therapy. American Journal of Hematology 2015;90:139–43. doi:10.1002/ajh.23877.
- Hulbert ML, Panepinto JA, Scott JP, Liem RI, Cook LJ, Simmons T, et al. Red blood cell transfusions during sickle cell anemia vaso-occlusive crises: a report from the magnesium in crisis (MAGiC) study. Transfusion 2017;57:1891–7. doi:10.1111/trf.14155.
- Mcgann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opinion on Drug Safety 2015;14:1749–58. doi:10.1517/14740338.2015.1088827.
- Agrawal RK, Patel RK, Shah V, Nainiwal L, Trivedi B. Hydroxyurea in Sickle Cell Disease: Drug Review. Indian Journal of Hematology and Blood Transfusion 2013;30:91–6. doi:10.1007/s12288-013-0261-4.
- Lucarelli G, Isgro A, Sodani P, Gaziev J. Hematopoietic Stem Cell Transplantation in Thalassemia and Sickle Cell Anemia. Cold Spring Harbor Perspectives in Medicine 2012;2. doi:10.1101/cshperspect.a011825.
- Angelucci E, Matthes-Martin S, Baronciani D, Bernaudin F, Bonanomi S, Cappellini MD, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica 2014;99:811–20. doi:10.3324/haematol.2013.099747.
- Brittenham GM. Iron-Chelating Therapy for Transfusional Iron Overload. New England Journal of Medicine 2011;364:146–56. doi:10.1056/nejmct1004810.
- Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood 2016;127:810–9. doi:10.1182/blood-2015-09-618553.
- Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 2015;125:2597–604. doi:10.1182/blood-2014-12-615948.
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood 2016;127:839–48. doi:10.1182/blood-2015-09-618587.
- Canver MC, Orkin SH. Customizing the genome as therapy for the β-hemoglobinopathies. Blood 2016;127:2536–45. doi:10.1182/blood-2016-01-678128.
- Soni S, Tisdale JF, Hsieh M, Kanter J, Leboulch P, Cavazzana M. Outcomes of Gene Therapy for Severe Sickle Cell Disease (SCD) via Transplantation of Autologous Hematopoietic Stem Cells Transduced Ex Vivo with a Lentiviral ßAT87Q-Globin Vector. Biology of Blood and Marrow Transplantation 2016;22. doi:10.1016/j.bbmt.2015.11.357.
Received: 11 April 2019
Accepted: 20May 2019
María Belén Paredes, María Eugenia Sulen
School of Biological Sciences and Engineering, Yachay Tech University
Corresponding author:[email protected],